Agence d'État Bulletin Officiel de l'État






Contenu non disponible en français
Ilustrísimos señores:
El conocimiento de las características del suelo cultivable es de la mayor importancia para la agricultura; pues permite realizar una fertilización correcta, lo cual constituye uno de los factores de producción más rentable.
Las diagnosis de carencias y las recomendaciones de fertilización deben basarse en resultados analíticos obtenidos mediante la aplicación de métodos de análisis uniformes.
Por otra parte, el conocimiento de la calidad tanto física como química y biológica de las aguas para los usos agrarios puede adquirirse a través de métodos de análisis normalizados, que en muchos casos coinciden con los métodos de análisis de suelos.
Teniendo en cuenta que la actuación de los distintos Organismos requiere con frecuencia la realización de análisis de idénticos componentes de los suelos y de las aguas, parece conveniente unificar criterios y aunar esfuerzos mediante el establecimiento de métodos oficiales de análisis únicos para este Ministerio.
Se ha considerado conveniente adaptar en lo posible la redacción de los citados métodos de análisis a los aprobados por Organismos Internacionales especializados en la materia, con el fin de aprovechar la experiencia habida en su aplicación y de facilitar la confrontación de los resultados analíticos En consecuencia, a propuesta de la Comisión de Métodos Oficiales de Análisis, creada por Orden ministerial de 5 de abril de 1972, este Ministerio acuerda:
Aprobar como oficiales los métodos de análisis de suelos y aguas, que se citan en el anexo único.
El Servicio de Publicaciones de este Ministerio publicará estos métodos de análisis que serán obligatorios para todos los laboratorios dependientes de este Ministerio.
Quedan derogadas las disposiciones de igual o inferior rango que se opongan a la presente Orden.
La presente disposición entrará en vigor a los treinta días de su publicación en el «Boletín Oficial del Estado».
Lo que comunico a VV. II.
Dios guarde a VV. II.
Madrid, 5 de diciembre de 1975.
OÑATE GIL
Ilmos. Sres. Subsecretario del Departamento, Directores generales de la Producción Agraria, Industrias y Mercados en Origen de Productos Agrarios, Capacitación y Extensión Agrarias, Servicio Nacional de Productos Agrarios, Instituto para la Conservación de la Naturaleza, Presidentes del FORPPA, INIA, IRYDA y Secretario general Técnico.
ABREVIATURAS, SIMBOLOS, DEFINICIONES Y NOTAS
| atm. | atmósfera. |
| C. | centígrado. |
| cm. | centímetro. |
| d. | densidad. |
| f. c. r. | fuerza centrífuga relativa. |
| g. | gramo. |
| h. | hora. |
| Ha. | hectárea. |
| Kg. | kilogramo. |
| l. | litro. |
| M. | molar, |
| m. | metro. |
| meq. | miliequivalente. |
| mg. | miligramo. |
| min. | minuto. |
| ml. | mililitro. |
| mm. | milímetro. |
| nm. | nanómetro. |
| N. | normal. |
| p. e. | punto de ebullición. |
| p. f. | punto de fusión. |
| pH | logaritmo cambiado de signo de la concentración de iones hidrógeno. |
| p. p m. | partes por millón. |
| p/p. | peso a peso. |
| p/V. | peso a volumen. |
| r. p. m. | revoluciones por minuto. |
| s. | segundo. |
| Tm. | tonelada métrica. |
| V/V. | volumen a volumen. |
| º | grado. |
| % | por ciento. |
| µ | micra. |
| µg | microgramo. |
| µl | microlitro. |
| ∑ | suma de. |
| / | dividido por. |
| × | multiplicado por. |
| < | menor que. |
| > | mayor que. |
| < > | equivalente a. |
| ≅ | aproximado a. |
Absorbencia: Logaritmo cambiado de signo de la relación de las transmitancias de la muestra y del material de referencia o patrón.
Estas unidades corresponden a las declaradas de uso obligatorio por la Ley 88/1967, de 8 de noviembre, y Decreto 1256/ 1974, de 25 de abril.
METODOS QUIMICOS
Suelos
1. PREPARACION DE LA MUESTRA
1.1. Principio
La muestra natural de un suelo, cuando llega al laboratorio, debe ser acondicionada como fase previa para la realización de los distintos análisis. Este acondicionamiento incluye la separación de los posibles elementos gruesos, la preparación de la muestra para análisis físicos y la preparación para ciertos análisis químicos.
1.2. Material y aparatos
1.2.1. Bandejas numeradas para colocar la muestra.
1.2.2. Tamiz de 2 mm. de luz.
1.2.3. Martillo de goma o madera, rodillo o molino especial para disgregar los terrones, con una separación entre muelas superior a 1 cm.
1.3. Procedimiento
Colocar la muestra en una bandeja y disgregar a mano, si es posible, los terrones existentes.
Mantener las bandejas al aire hasta que se equilibre su humedad con la del laboratorio. Durante la desecación, disgregar a mano los terrones existentes. El desprendimiento de polvo es un indicio de haber logrado este equilibrio.
Pesar la muestra con la aproximación del gramo.
Disgregar mecánicamente los terrenos mediante un martillo de goma o madera, rodillo o molino especial.
Tamizar la totalidad de la muestra por un tamiz de 2 milímetros de luz.
Recoger la porción que haya pasado por el tamiz en un recipiente.
Esta fracción se denomina tierra fina seca al aire (TFSA).
Efectuar un nuevo tamizado con la fracción que no pasó por el tamiz en una pileta y con ayuda de agua hasta observar que los elementos gruesos quedan limpios.
Secar los elementos gruesos en estufa, dejar que se enfríen al aire y pesarlos con aproximación del gramo.
Conservarlos para posibles futuros análisis.
1.4. Cálculo


p’ = peso, en gramos, de los elementos gruesos
p = peso, en gramos, de la muestra.
1.5. Observaciones
Para la determinación del potasio asimilable se aconseja no secar la muestra al aire.
1.6. Referencias
1. Soils Bulletin núm. 10 FAO phisical and chemical Methods and Water analysis Pág. 19 Roma, 1970.
2. Buebs, R. E. G. Stanford and A D. Scott (1956). Relation of avaible potassion to soil moisture. Soil Sci. Soc. Amer. Proc. 20: 45-50.
2. pH
2.1. Principio
Medida del potencial eléctrico que se crea en la membrana de vidrio de un electrodo, que es función de las actividades de iones hidrógeno a ambos lados de la membrana, utilizando como referencia un electrodo de calomelanos con puente salino.
2.2. Material y aparatos
2.2.1. Potenciómetro (pH metro) y juego de electrodos de vidrio y calomelanos.
2.2.2. Vasos de 100 ml.
2.2.3. Varillas agitadoras o agitador magnético.
2.3. Reactivos
2.3.1. Solución CIK 0,1 M. Disolver 7,456 g. de CIK en 100 mililitros de agua destilada y diluir hasta 1 litro.
2.3.2. Solución tampón de biftalato potásico 0,05 M. Secar la sal durante dos horas a 110 C. Disolver 10,21 g. de sal en agua destilada y diluir hasta 1 litro. Como conservador añadir a la solución tampón 1 ml. de cloroformo o un cristal de timol de unos 10 mm. de diámetro. Esta solución tiene un pH de 4,00 en el intervalo de temperatura de 15° a 30° C.
2.3.3 Solución tampón de PO4H2K 0,025 M y PO4HNa2 0,025 M. Secar las dos sales durante dos horas a 110° C. Disolver 3,44 gramos de PO4H2K y 3,55 g. de PO4HNa2 en agua destilada y diluir hasta 1 litro. Como conservador, añadir 1 ml. de cloroformo o un cristal de timol de unos 10 mm. de diámetro a la solución tampón. Esta solución tiene un pH de 6,90 a 15° C, de 6,88 a 20° C, de 6,86 a 25° C y de 6,85 a 30° C.
2.4. Procedimiento
2.4.1. pH en agua: Pesar 10 g. de suelo y añadir 25 ml. de agua destilada. Agitar vigorosamente con varilla o agitador magnético. Dejar reposar durante treinta minutos. Ajustar la posición de los electrodos en el soporte, de manera que, cuando desciendan en el vaso, el electrodo de vidrio se sumerja bien en la parte parcialmente sedimentada de la suspensión y el electrodo de calomelanos quede en la solución-suspensión sobrenadante para que se establezca buen contacto eléctrico a través del capilar del puente salino. Agitar la suspensión inmediatamente antes de introducir los electrodos, pero no durante la medida. Medir el pH de acuerdo con las instrucciones específicas del potenciómetro, utilizando una solución tampón de pH próximo al del suelo.
2.4.2. pH en cloruro potásico. Proceder igual que en 2.4.1, utilizando el reactivo (2.3.1) en vez de agua destilada.
2.5. Expresión de los resultados
Expresar los resultados de 2.4 (1 y 2) como:
«pH medido en suspensión suelo: agua 1: 2,5 y pH medido en suspensión suelo: solución CIK 0,1 M 1: 2,5, respectivamente.»
2.6. Observaciones
2.6.1. Para pH superior a 9 debe utilizarse un electrodo de vidrio especial.
2.6.2. Cuando no se obtenga una suspensión fluida con la relación suelo: agua indicada (caso de suelos con alto contenido en materia orgánica) determinar el pH en la pasta saturada, al cabo de dos horas de haberla preparado.
2.7. Referencia
1. Chapman, H. D., y Pratt, P. F.: «Methods of Analysis for Soil Plants and Water», 233-234. University of California. División of Agricultural Sciences, 1961.
3 (a) CARBONATOS
3(a).1. Principio
Tratando los carbonatos con ácido en un dispositivo cerrado, a presión y temperatura constante, el incremento de volumen es una medida directa del CO2 desprendido cuando no se produzcan otros gases.
Este método no es aplicable a suelos con contenido muy altos de materia orgánica o con cantidades apreciables de MnO2. En presencia de dolomía, ver 3(a).6.3.
La interferencia potencial causada por MnO2 puede minimizarse usando un ácido que contenga un agente reductor y la causada por altos contenidos de materia orgánica reduciendo el tiempo de reacción o bien destruyéndola previamente.
3(a).2. Material y aparatos
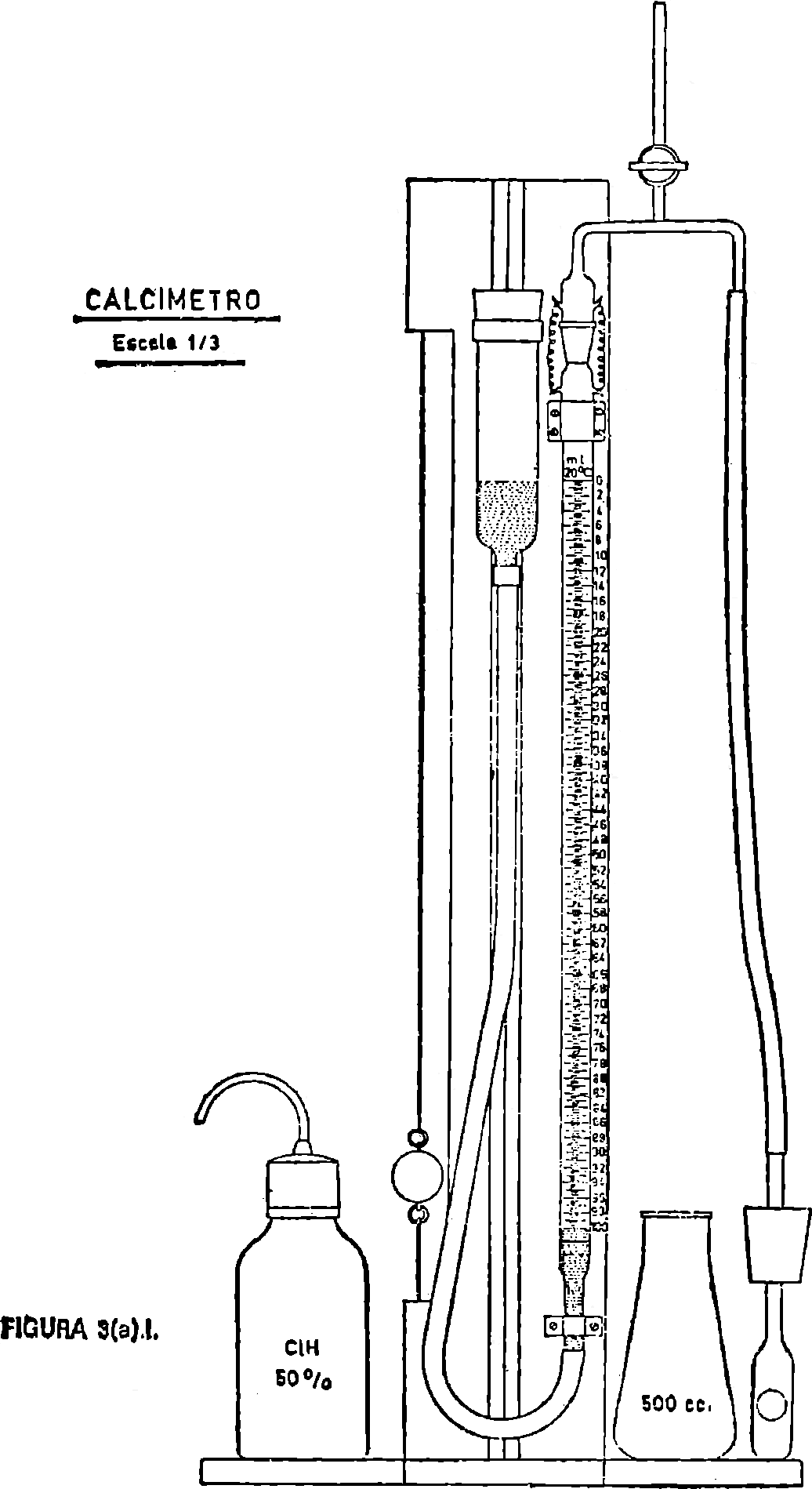
3(a).2.1. Calcímetro según la figura 3(a).1 o similar con cierres herméticos.
3(a).2.2. Matraces erlenmeyer.
3 (a).3. Reactivos
3(a).3.1. Acido clorhídrico 1:1 (aprox. 6 N).
3(a).3.2. Solución saturada de ClNa y CO2. Disolver 100 g. de ClNa y 1 g. de CO3HNa en 350 ml. de agua; a esta solución agregar lentamente ácido sulfúrico diluido hasta reacción ácida al rojo de metilo, agitando para eliminar el exceso de C02. Esta solución coloreada en rojo por el indicador se utiliza para llenar el calcímetro.
3(a).3.3. Carbonato cálcico finamente pulverizado.
3 (a).4. Procedimiento
Pesar de 0,2 a 5 g. de tierra fina, previamente triturada, dependiendo del ensayo previo efectuado y colocarla en un matraz de 500 cm3; humedecer la muestra con unos pocos cm3 de agua y conectar el erlenmeyer al calcímetro, en el que previamente se habían colocado unos ml. de ácido clorhídrico al 50 por 100 (reactivo 3(a).3.1), usando el dispositivo al efecto. Con la llave del calcímetro abierta para mantener en el interior del sistema la presión atmosférica, ajustar la altura del depósito del calcímetro hasta enrasar la bureta del mismo con el 0. Cerrar la llave, e inclinando el erlenmeyer, verter el ácido sobre la muestra, agitando suavemente para favorecer el ataque. Al mismo tiempo se va descendiendo la rama móvil del calcímetro, procurando mantener un mismo nivel del líquido en las dos ramas.
Cuando el nivel del líquido del calcímetro permanezca estacionario, dejar de agitar y tomar la lectura alcanzada por el mismo una vez enrasadas las dos ramas. El volumen leído corresponde al del C02 desprendido por la muestra.
Repetir las mismas operaciones tomando en vez de suelo, 0,2 g. de CO3Ca (reactivo 3(a).3.3), y con las lecturas obtenidas, efectuar los cálculos.
3 (a).5. Cálculos

Siendo:
L = lectura observada en el calcímetro para la muestra.
L' - lectura observada en el calcímetro para el CO3Ca.
P = peso seco, en g. de la muestra de suelo.
P' = peso, en g., de CO3Ca (0,2 según el método descrito).
3(a).6. Observaciones
3(a).6.1. Para agitar el erlenmeyer debe usarse unas pinzas para evitar modificar la temperatura con el calor de la mano.
3(a).6.2. Para evitar las interferencias producidas por la presencia de MnO2 debe modificarse el reactivo 3(a).3.1, agregándole 3 g. de Cl2Fe.4H2O por cada 100 ml. inmediatamente antes de usarlo.
3 (a).6.3. En el caso de suelos con carbono cálcico-magnésico (dolomía) debe utilizarse el método 3(b).
3(a).7. Referencias
1. Demolon, A., y Leroux, D.: «Guide pour l’étude experimental des sois». Gauthier Villars. París. 1952.
2. Allison, L. E., y Moodie, C.-D.: «Methods of Soil. Analysis». Part 2, 1389-1392. American Society of Agronomy, 1965.
3 (b) CARBONATOS
(Volumetría) (Provisional)
3(b).1. Principio
Los carbonatos son atacados por ácido perclórico y el exceso de ácido se valora con hidróxido sódico.
3(b).2. Material y aparatos
3(b).2.1. Matraces erlenmeyer de 250 ml.
3(b).2.2. Agitador magnético y barras agitadoras.
3.(b).2.3. Potenciómetro (pH-metro) y electrodo combinado de vidrio y calomelanos.
3(b).2.4. Buretas.
3(b).2.5. Placa calefactora.
3(b).3. Reactivos
3(b).3.1. Acido perclórico 1N. Aforar 86,2 ml, de HClO4 70 por 100 a un litro. Normalizar esta solución, cada vez que se emplee, valorando 10 ml. de la misma frente al reactivo 3(b).3.2.
3(b).3.2. Hidróxido sódico 0,5N. Aforar a un litro una ampolla de solución concentrada a tal efecto.
3(b).3.3. Anaranjado de metilo al 0,1 por 100 en agua.
3(b).4. Procedimiento
Pesar 0,5 g. de tierra triturada y colocarla en un matraz erlenmeyer de 250 ml. Añadir 10 ml. del reactivo 3(b).3.1. Hervir un minuto en placa calefactora y dejar enfriar. Añadir 150 ml. de agua destilada e introducir en el interior del matraz una barra agitadora con recubrimiento de teflón.
Valorar con 3(b).3.2. mediante bureta, utilizando el agitador magnético y con el potenciómetro incorporado. Añadir el reactivo rápidamente hasta pH = 3,5 y gota a gota hasta pH = 6,5, punto final de la valoración. Cuidar siempre de ajustar el aparato con una solución tamponada de referencia.
3(b).5. Cálculos
Calcular el contenido en carbonatos, expresado en porcentaje de caliza, del suelo.

M = miliequivalentes añadidos de ácido perclórico.
M' = miliequivalentes gastados de hidróxido sódico.
P = peso en gramos de la muestra.
3(b).6. Observaciones
3(b).6.1. Es preciso evitar que, por descuido, el matraz con el perclórico quede seco al hervir un minuto, ya que puede existir peligro de explosión.
3(b).6.2. Si se dispone de potenciógrafo automático, la valoración con hidróxido sódico es mucho más cómoda y los resultados son más exactos y reproducibles al aparecer perfectamente definido el punto de equivalencia.
3(b).7. Referencias
1. Jackson, M. L.: «Análisis químico de suelos». Omega. Barcelona, 1964.
2. Allison, L. E., y Moodie, C. D.: «Method of Soils Analysis». Part 2 (1379-1396). American Society of Agronomy, 1965.
3. Hesse, P. R.: «A Texbook of Soil Chemical Analysis». John Murray. London, 1971.
4. FOSFORO SOLUBLE EN BICARBONATO SODICO
(Provisional)
4.1. Principio
Se extrae el fósforo del suelo con CO3 H Na 0,5 M a un pH aproximadamente constante de 8,5. En los suelos calizos, alcalinos o neutros que contengan fosfatos cálcicos, aumenta la concentración de P en la solución extractora por precipitación de Ca como CO3 Ca.
En los suelos ácidos que contengan fosfatos de hierro y aluminio, la concentración de P en la solución aumenta conforme el pH se eleva.
4.2. Material y aparatos
4.2.1. Fotocolorímetro.
4.2.2. Papel de filtro Whatman núm. 40.
4.3. Reactivos
4.3.1. Solución de bicarbonato sódico (CO3 H Na), 0,5 M. Ajustar el pH de esta solución a 8,5 con Na OH 1 M. Añadir aceite mineral para evitar el contacto de la solución con el aire. Preparar una solución fresca antes de usarla si ha estado más de un mes en un frasco de vidrio. Se preservará mejor en envase de polietileno, pero comprobar el pH de la solución todos los meses.
4.3.2. Carbón activado. Usar preferentemente carbón negro G.
4.3.3. Solución de molibdato amónico (Mo7 O24 (NH4)6 4 H2O). Disolver 15 g. de Mo7 O24 (NH4)6 4 H2O en 300 ml. de agua destilada templada. Filtrar la mezcla si fuese necesario y dejar enfriar. A continuación añadir 342 ml. de Cl H concentrado poco a poco y mezclando conforme se añada. Diluir hasta un litro con agua destilada. Esta solución contiene 50 ml. de exceso de Cl H concentrado para neutralizar el CO3 H Na contenido en 5 ml. de alícuota.
4.3.4. Solución concentrada de cloruro estannoso (Cl2 Sn 2H2O). Disolver 10 g. de Cl2 Sn 2H2O en 25 ml. de Cl H concentrado. Preparar solución nueva al menos cada dos meses. Utilizar cristales grandes de la sal mejor que polvo fino. Conservar la solución en atmósfera de hidrógeno generada por Zn y ClH o en un refrigerador.
4.3.5. Solución diluida de cloruro estannoso. Añadir 0,5 ml. de solución concentrada Cl2 Sn 2H2O (reactivo 4.3.4) a 66 ml. de agua. Preparar esta solución para cada serie de determinaciones.
4.3.6. Solución patrón de fósforo. Pesar 0,4393 g. de fosfato de potasio (PO4 H2 K) y verter en un matraz aforado de un litro. Añadir 500 ml. de agua destilada y agitar hasta que se disuelva la sal. Diluir la solución hasta un litro con agua. Añadir cinco gotas de tolueno para disminuir la actividad microbiana. Esta solución contiene 0,1 mg. de P por ml.
4.3.7. Solución diluida de fósforo. Diluir con agua 20 ml. de la solución patrón de fósforo (reactivo 4.3.6) hasta un litro. Esta solución contiene 2 μg de P por ml.
4.4. Procedimiento
4.4.1. Extracción del P. Añadir a 5 g. de suelo una cucharilla de carbón (reactivo 4.3.2) y 100 ml. de solución extractora (reactivo 4.3.1) en un erlenmeyer de 250 ml. Agitar el matraz durante media hora. Filtrar la suspensión a través de un papel de filtro Whatman núm. 40. Añadir más carbón cuando sea necesario para obtener un filtrado claro. Agitar el matraz inmediatamente antes de verter la suspensión en el embudo con papel de filtro.
4.4.2. Determinación del P. Colocar una alícuota de 5 ml. de extracto en un matraz aforado de 25 ml. Añadir poco a poco 5 ml. de la solución de molibdato amónico (reactivo 4.3.3) directamente en el matraz. Una vez que el desprendimiento rápido de CO2 haya terminado, agitar el matraz suavemente para mezclar el contenido. Lavar el cuello del matraz con agua destilada para evitar el contacto directo de la solución de Cl2 Sn con la solución concentrada de molibdato amónico. Añadir 1 ml. de la solución diluida de Cl2 Sn (reactivo 4.3.5) al matraz y completar el volumen con agua, mezclando inmediatamente. Medir la longitud de onda de 660 mμ la transmitancia de la solución en el fotocolorímetro diez minutos después de la adición de la solución de Cl2 Sn.
Para preparar la curva estándar, añadir con pipeta fracciones alícuotas de la solución diluida de P (reactivo 4.3.7) que contenga de 2 a 25 μg de P en matraces aforados de 25 ml. A continuación añadir a cada matraz 5 ml, de la solución de CO3H Na (reactivo 4.3.1). Finalmente se desarrolla el color en la misma forma indicada para la solución problema. En papel semilogaritmico de un ciclo se representa el porcentaje de transmitancia respecto a la concentración de P.
4.5. Cálculo
Calcular el fósforo expresado en p. p. m. del suelo
Fósforo p. p m = 4P
P = peso en μg de fósforo en 5 ml. de la solución extractora.
4.6. Observaciones
4.6.1. No se deben de preparar patrones de concentración inferior a 0,08 μg/ml. por no desarrollarse en ellos el color azul típico. Cuando el P extraído por este método se encuentra en concentración inferior al patrón más diluido debe de aumentarse la cantidad inicial de suelo hasta 40 g. si fuese necesario.
4.6.2. Orientación de nivel crítico.
La relación entre el P soluble en el extracto y la respuesta de las cosechas a la fertilización fosforada es como sigue:
P < 5 p. p. m. respuesta.
P ≤ 10 p. p. m. respuesta probable.
P > 10 p. p. m. respuesta improbable.
Las cosechas con las que se han fijado esos niveles fueron trigo, avena, alfalfa y algodón.
4.7. Referencia
1. Olsen, S. R.; Cole, C. V.; Watanabe, F. S., y Dean, L. A.: «Estimation of available phosphorus in soils by extraction with sodium bicarbonate». U. S. Department of Agriculture». Circular 939. 1954.
5. POTASIO EXTRAIDO POR ACETATO AMONICO
5.1. Principio
El índice de asimilabilidad de K en suelos más universalmente usado es la suma del K intercambiable y del K soluble en agua, esto es, el K total extraído por una solución neutra 1 N de acetato amónico.
5.2. Material y aparatos
5.2.1. Fotómetro de llama o espectrofotómetro con accesorios para fotometría de llama.
5.2.2. Agitador mecánico de tubos de centrífuga.
5.2.3. Centrífuga y tubos de 50 ml.
5.3. Reactivos
5.3.1. Solución de acetato amónico (Ac O NH4) 1 N ajustada a pH 7. Por cada litro de solución que se prepare, añadir 57 ml. de ácido acético glacial a unos 600 ml. de agua, y sobre ello 68 ml. de NH4 OH concentrado, de peso específico 0,90. El NH4 OH debe de añadirse en una vitrina de gases a través de un embudo de cuello largo de tal manera que llegue al fondo de la solución del ácido. Dejar enfriar y ajustar a pH 7 con ácido acético o NH4 OH, usando un pH-metro o indicador azul de bromotimol. Diluir la solución al volumen convenido, mezclarla y almacenarla en un frasco pyrex.
5.3.2. Solución patrón de potasio. Disolver en agua 0,9533 gramos de Cl K desecado, diluir la solución a 500 ml. y mezclar. Esta solución contiene 1.000 p. p. m. de K.
5.4. Procedimiento
5.4.1. Extracción del K. Colocar 10 g. de suelo (5 g. si el suelo contiene más de 500 p. p. m. de K extractable) en un tubo de centrífuga de 50 ml. Añadir 25 ml. de Ac ONH4 (reactivo 5.3.1) y agitar durante diez minutos. Centrifugar el tubo hasta que el líquido sobrenadante esté claro. Decantar el líquido sobrenadante en un matraz de 100 ml. Hacer tres extracciones similares de igual manera. Enrasar hasta la marca de 100 ml. con Ac ONH4 y mezclar.
5.4.2. Determinación del K. A partir de la solución patrón de K. Determinar el contenido de K en el extracto de Ac ONH4 Ac ONH4 (reactivo 5.3.1) que contenga 0, 5, 10, 20, 40 y 60 p. p. m. de K. Determinar el contenido de K en el extracto de Ac ONH4 del suelo, comparando con la emisión producida por las soluciones patrones.
5.5. Cálculo
Calcular el K expresado en p. p. m. del suelo
Potasio, p. p. m. = 100 ×/m
x = contenido en K de la solución de Ac ONH4 expresado en p. p. m.
m = peso seco en g. de la muestra del suelo.
5.6. Observaciones
5.6.1. El extracto de Ac ONH4 del suelo puede ser pulverizado en un fotómetro de llama siempre que se evite la presencia de partículas sólidas que podrían ocluir el pulverizador. Si existen partículas sólidas en el extracto, debe filtrarse una parte del mismo usando papel de filtro Whatman núm. 40, antes de colocar la solución en el fotómetro de llama.
5.6.2. Cuando en la muestra de suelo se determinen los cationes intercambiables se utilizará para esta determinación el extracto de suelo tal como se describe en el Método 10.
5.6.3. Orientación de nivel crítico.
Dependiendo de la cosecha y del suelo se han citado cifras de niveles críticos de K comprendidas entre 50 y 150 p. p. m. Cualquier valor inferior o superior al definido por ese intervalo debe ser considerado, en principio, como deficiente y satisfactorio, respectivamente.
5.7. Referencia
1. Pratt, P. F.: «Methods of Soil Analysis». Part 2, 1027-1028. American Society of Agronomy, 1985.
6. PRUEBA PREVIA DE SALINIDAD. EXTRACTO SUELO AGUA. 1/5
6.1. Principio
Con este método se obtiene una evaluación grosera de la salinidad del suelo, evitándose la innecesaria y laboriosa preparación de la pasta saturada de suelo y subsecuente separación del extracto en muestras no salinas (método 13).
6.2. Material y aparatos
6.2.1. Agitador mecánico.
6.2.2. Frascos de cristal de 100 ml.
6.2.3. Embudos y tubos de ensayo.
6.3. Reactivos
6.3.1. Solución de hexametafosfato sódico al 0,1 por 100. Disolver 0,1 g. de hexametafosfato sódico en agua y diluir en 100 ml.
6.4. Procedimiento
Pesar en un granatario 10 g. de suelo desecado al aire y colocarlo en un frasco. Añadir 50 ml. de agua destilada y tapar el frasco. Agitar en el agitador mecánico durante media hora. Si no se dispone de agitador, agitar el frasco a mano vigorosamente durante un minuto al menos cada quince minutos para un tiempo total de una hora. Dejar reposar unos minutos y filtrar la suspensión a través de un papel de filtro de alto poder retentivo y bajo contenido en cenizas. Desechar el filtrado inicial en caso de ser turbio. Añadir una gota de hexametafosfato sódico al 0,1 por 100 en los 25 ml. de filtrado que se hayan recogido en un tubo de ensayo.
Determinar la conductividad eléctrica del extracto acuoso tal como se describe en el método 7.
6.5. Cálculo
Como en 7.5.
6.6. Observaciones
Cuando la salinidad en el extracto 1:5 sea superior a 0,2 mmhos/cm. se preparará la pasta saturada de suelo y el correspondiente extracto se separará para el estudio de la salinidad (ver método 13). El hexametafosfato sódico se añade al extracto para evitar la precipitación de CO3Ca. La cantidad de hexametafosfato sódico añadida aumenta la concentración de Na en la solución en menos de 0,5 p. p. m., lo cual es despreciable comparado con la precipitación de CO3Ca.
6.7. Referencia
1, Bower, C. A., y Wilcox, L. V.: «Methods of Soil Analysis». Part 2, 935-936. American Society of Agronomy, 1965.
7. CONDUCTIVIDAD ELECTRICA
7.1. Principio
El agua que contiene sales disueltas del tipo que normalmente se encuentran en el suelo conduce la corriente aproximadamente en proporción a la cantidad de sal disuelta. Por consiguiente, la medida de la conductividad eléctrica de un extracto da una indicación de la concentración total de los constituyentes ionizados.
La determinación de la conductividad eléctrica se hace midiendo la resistencia eléctrica entre dos electrodos paralelos sumergidos en la solución acuosa.
Como la conductividad eléctrica de las soluciones acuosas de sales aumenta con la temperatura, se refiere a 25° C.
7.2. Material y aparatos
7.2.1. Puente de Wheatstone del tipo de corriente alterna con 1.000 a 50 ciclos con un indicador catódico del tipo ojo mágico. Existe en el mercado un puente de Wheatstone con celda de conductividad de una constante 0,5 cm-1, especialmente diseñado para medida de conductividad eléctrica en extractos de saturación de suelos. La compensación de temperatura adecuada hace que en la escala que abarca de 0,15 a 15 mmhos/cm. se pueda leer directamente la conductividad de la solución a 25° C.
7.2.2. Celda de conductividad, del tipo pipeta, con electrodos de platino platinizados. La constante de la celda debe ser aproximadamente 1,0 cm-1. Cuando se analizan suelos muy salinos es deseable tener otra celda de conductividad con una constante de unos 10,0 cm-1.
Lavar las celdas nuevas con solución limpiadora de ácido crómico y sulfúrico y platinizarlas antes de usarlas. Lavar y replatinizar la celda cada vez que las lecturas sean erróneas o defectuosas o cuando el platino se haya desprendido total o parcialmente. Preparar la solución platinizante disolviendo 1 g. de cloruro de platino y 0,012 g. de acetato de plomo en 100 ml. de agua. Para platinizar los electrodos llenar la celda con la solución anterior y conectar los terminales de los electrodos a los bornes de una batería de 1,5 voltios, haciendo pasar la corriente a través de la celda. Intercalar un shunt en la batería cuando sea necesario ajustar la corriente, de manera que se desprenda una pequeña cantidad de gas. Dar por terminado el proceso cuando los electrodos estén cubiertos con un depósito de negro platino. Drenar la celda y recuperar para el futuro uso la solución platinizante. Enjuagar bien la celda con agua destilada y dejarla llena de agua destilada cuando no se use.
7.3. Reactivos
7.3.1. Solución de cloruro potásico, 0,01 N. Disolver 0,7456 gramos de Cl K en agua destilada y añadir agua hasta completar un litro a 25.º C. Esta es la solución patrón de referencia. A 25.º C tiene una conductividad eléctrica de 0,0014118 mho/cm.
7.4. Procedimiento
Dejar que la solución patrón de Cl K 0,01 N y las muestras de aguas o extractos acuosos del suelo estén a la misma temperatura. Cualquier temperatura en el intervalo de 20° a 30.º C es aceptable, pero es importante que las soluciones problemas y la patrón estén a la misma temperatura. Cuando se requiera gran precisión se colocarán las soluciones problemas y la patrón en un termostato o una cámara de temperatura constante a 25° C. Enjuagar y llenar la celda de conductividad con la solución patrón. Equilibrar el puente de Wheatstone según se indique en el manual de instrucciones y anotar la resistencia de la celda (R) en ohmios. Enjuagar y llenar la celda con la solución problema. Si el volumen de ésta es limitado, enjuagar la celda con agua destilada y seguidamente con acetona, y secarla al aire hasta que la acetona se evapore. Dejar que la celda alcance la temperatura de las soluciones. Equilibrar el puente y anotar la resistencia de la celda R’ en ohmios.
7.5. Cálculo
Calcular la conductividad eléctrica de la solución problema en mhos/cm. a 25°.

0,0014118 = conductividad eléctrica de la solución 0,01 N de ClK en mhos/cm. a 25°.
R = resistencia en ohmios de la celda con la solución patrón.
R' = resistencia en ohmios de la celda con la solución problema.
Para expresar la conductividad eléctrica en mmhos/cm. a 25° se multiplica el resultado anterior por 1.000.
Expresar los resultados de conductividad eléctrica cuando sean inferiores a 1 con dos cifras decimales, y si son superiores a 1 con tres cifras significativas.
7.6. Observaciones
7.6.1. La medida de la conductividad eléctrica es precisa y reproductible hasta tres cifras significativas en el intervalo que se encuentra normalmente en los extractos de suelo saturado.
7.6.2. Existe una alta correlación positiva entre la conductividad eléctrica, la concentración total de cationes y aniones y la presión osmótica de los extractos de suelo saturado. Debido a las marcadas diferencias de pesos equivalentes y conductividades equivalentes entre los diversos constituyentes, la correlación existente entre la conductividad eléctrica y la concentración de sales en porcentaje de peso es algo más pobre. Los siguientes factores de conversión aproximados se manejan frecuentemente.
Concentración de sal, mg/1. = 640 x CE en mmho/cm.
Concentración total de cationes, meq/l. = 10 x C E en mmho/cm.
Presión osmótica, atm. = 0,36 x C E en mmho/cm.
7.7. Referencias
1. Bower, C. A., y Wilcox, L. V.: «Methods of Soil Analysis». Part 2, 937-940. American Society of Agronomy, 1965.
2. National Research Council, 1929. International Critical Tables.
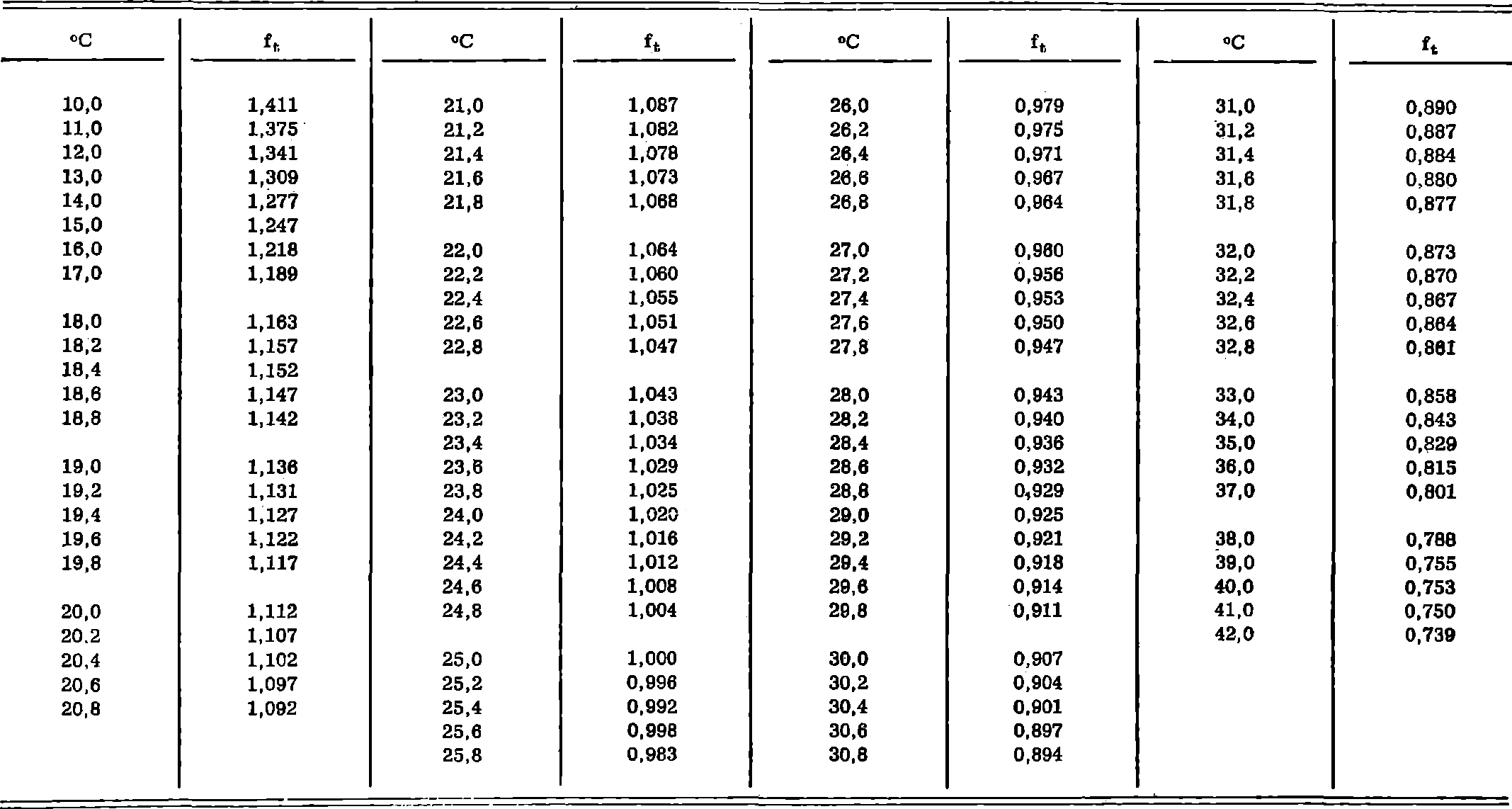
TABLA 7.I.
Factores de temperatura para corregir los datos de resistencia y conductividad de soluciones acuosas a la temperatura de 25° C
CE25 = CEt × ft : R25 = Rt/ft
8(a) NITROGENO ORGANICO
8(a).1. Principio
Con este método se determina el nitrógeno constituyente de la materia orgánica del suelo más el nitrógeno de la fracción mineral circunstancialmente en forma de ion amónico. Es una de la muchas variantes del método Kjeldahl (macro) que se usa en análisis de suelos.
El método 8(a) se recomienda especialmente para aquellos estudios en los que se requiera conocer la relación C/N de la materia orgánica del suelo, y también en muestras de suelos en las que interesando conocer el N total tengan muy bajo contenido de N bajo la forma de nitrato o nitrito.
El contenido total de N en los suelos varía entre menos de 0,02 por 100 en los subsuelos a 2,5 por 100 en turbas. La capa arable de la mayoría de los suelos cultivados contiene entre 0,06 y 0,5 por 100 de N.
8(a).2. Material y aparatos
8(a).2.1. Frascos de digestión macro-Kjeldahl (350 ó 500 ml).
8(a).2.2. Batería de digestión.
8(a).2.3. Aparatos de destilación macro-Kjeldahl (frascos de 800 ml.).
8(a).2.4. Erlenmeyer de 500 ml.
8(a).2.5. Microbureta de 25 ml.
8(a).2.6. Frasco para conservar reactivo 8(a).3.5. [fig. 8(a).I].
8(a).3. Reactivos
8(a),3.1. Acido sulfúrico concentrado.
8(a).3.2. Sulfato potásico.
8(a).3.3. Sulfato de cobre. (SO4Cu 5 H2O)
8(a).3.4. Selenio.
8(a).3.5. Hidróxido sódico, aproximadamente 10 N.
Cuando se vayan a realizar muchas determinaciones conviene preparar el reactivo de la siguiente forma: Se empieza por preparar una disolución al 50 por 100 de Na OH y se deja que se sedimenten sus impurezas. Para hacer esto, disolver 3 Kg. de Na OH en 3 litros de agua en un frasco de vidrio pyrex de paredes gruesas. La disolución así preparada se deja en reposo durante varios días con el fin de que se sedimente el CO3 Na2. A continuación se separa con precaución y mediante un sifón, la solución clara que sobrenada, dejándola caer sobre 1,5 l. de agua. Mezclar la solución y colocarla en el frasco destinado a guardar el reactivo del Na OH al 40 por 100 [ver figura 8(a).I].
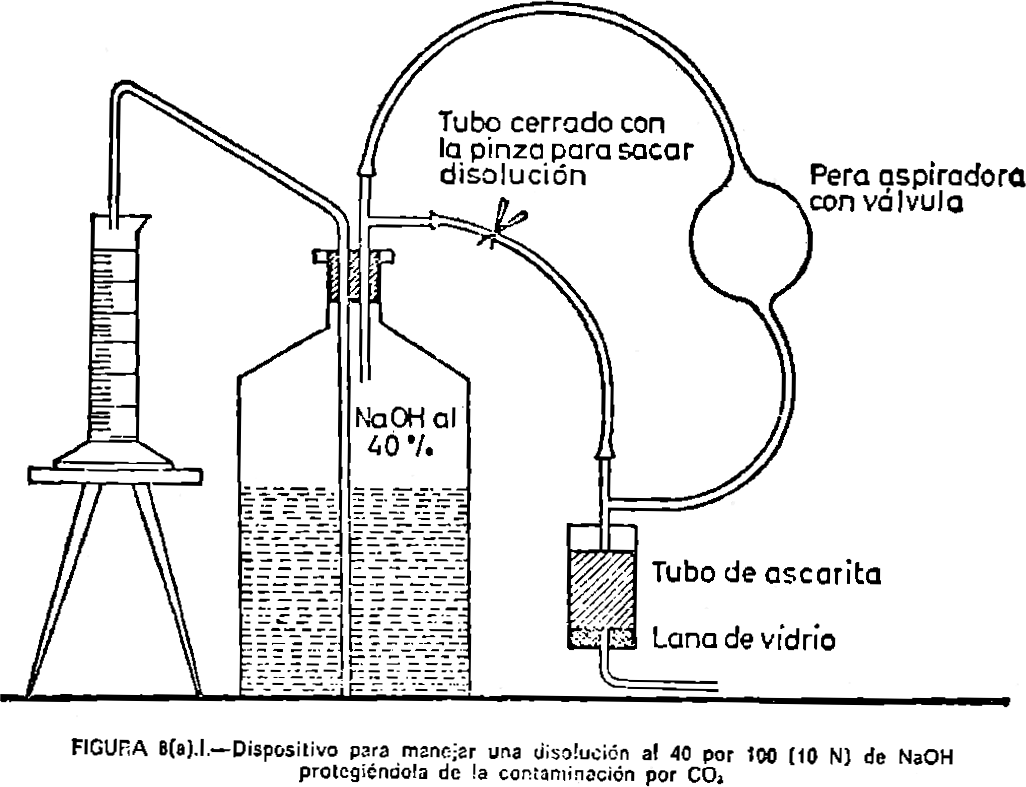
8(a).3.6. Solución mezcla de ácido bórico-indicador. Colocar 80 g. de ácido bórico puro (B O3 H3) en un frasco de 5 litros con la marca de 4 litros y añadir 3,8 litros de agua. Calentar y agitar el frasco hasta que se disuelva el ácido. Enfriar la solución y añadir 80 ml. de la solución de indicadores. Esta solución se prepara disolviendo 0,099 g. de verde de bromocresol y 0,066 g. de rojo de metilo en 100 ml. de etanol. Entonces, añadir con cuidado Na OH al frasco de 5 litros hasta que la solución tome un tinte morado rojizo (pH aprox. 5.0) y después añadir agua hasta completar 4 litros. Mezclar la solución a fondo antes de usarla.
8(a).3.7. Acido sulfúrico (o clorhídrico), 0,05 N.
8(a).4. Procedimiento
Colocar una muestra que contenga unos 10 mg. de N (1 g. de turba, 5 ó 10 g. de suelo y 20 g. si es muy arenoso) que haya pasado a través de un tamiz de 1 mm. de abertura en un matraz seco macro-Kjeldahl y añadir 20 ml. de agua. Después de girar el matraz durante unos minutos, dejarlo en reposo durante unos treinta minutos. Añadir entonces 10 g. de SO4 K2, 1 g. de SO4 Cu 5 H2O, 0,1 g. de Se y 30 ml. de SO4 H2 concentrado y calentar el matraz con cuidado en batería de digestión. Cuando el agua se haya eliminado y no aparezca más espuma, aumentar el calentamiento hasta que el líquido de digestión aparezca claro. Entonces, hervir la mezcla durante cinco horas. Regular el calor durante este período de ebullición de tal manera que el SO4 H2 se condense en el tercio inferior del cuello del matraz.
Finalizada la digestión, dejar que se enfríe el matraz y añadir unos 100 ml. de agua (despacio y agitando). Enfriar el matraz con chorro de agua fría y transferir el contenido a un matraz de destilación macro-Kjeldahl. Retener el residuo arenoso en el matraz de digestión en la medida que sea posible, ya que la arena puede entorpecer la destilación. Cuatro lavados del residuo arenoso con 50 ml. de agua suelen ser suficientes para transferir cuantitativamente el amonio en el liquido de digestión.
Para determinar el N amónico liberado en la digestión, colocar un erlenmeyer de 500 ml. conteniendo 50 ml. del reactivo 8(a).3.6. debajo del condensador, de manera que el tubo del último quede introducido en la solución de ácido boricoindicador. Colocar el frasco de destilación a un ángulo de 45°. añadir una cucharilla de piedra pómez y verter 150 ml. de Na OH 10 N que se deslice por la pared del frasco sin que se mezcle apreciablemente con el líquido de digestión. Conectar rápidamente el frasco al aparato de destilación, mezclar el contenido a fondo agitando e inmediatamente comenzar la destilación. Regular el calentamiento para evitar la ascensión del ácido bórico a través del tubo de condensación y para minimizar la formación de espuma y proyección de sólidos en el interior del frasco durante la destilación. Comprobar que el caudal de agua fría que atraviesa el condensador es suficiente para mantener la temperatura del destilado por debajo de 35° C. Cuando se hayan destilado unos 150 ml., bajar el erlenmeyer de manera que el extremo del tubo del condensador esté por encima de la superficie del líquido de destilación. Enjuagar el extremo del condensador con agua y dar por terminada la destilación.
Determinar el amonio en el líquido de destilación, valorándolo con SO4 H2 0,05 N en una bureta graduada a intervalos de 0,1 ml. La mezcla de indicadores es verde en medio alcalino y roja en medio ácido. El cambio de color del viraje es de verde a rosa.
8(a).5. Cálculo
Calcular el N orgánico del suelo referido a porcentaje

V = volumen en ml. de SO4 H2 0,05 N utilizado.
P = peso en g. de la muestra referido a materia seca.
8(a).6. Observaciones
La rejilla con asbesto para, el matraz de digestión es necesaria para evitar que la llama esté en contacto directo con el matraz por encima del nivel de la mezcla de ácidos.
8(a).7. Referencia
1. Bremner, J. M.: «Methods of Soil Analysis», Part 2. 1.162-1.164. American Society of Agronomy, 1965.
8(b) NITROGENO (ORGANICO + NITRATO)
8(b).1. Principio
Es una modificación del método 8(a) para reducir los nitratos a amonio previamente a la destilación del líquido de digestión.
Se recomienda este método para el análisis del N total de los suelos con contenido de nitrato apreciable y de nitritos despreciable. En caso de interesar la determinación del N total que incluya nitritos, se recomienda usar la modificación de Olsen (1929) al método macro-Kjeldahl tal como es descrita por Bremner (1965).
8(b).2. Material y aparatos
Como en 8(a).2.
8(b).3. Reactivos
Como en 8 (a).3 (1, 2, 3, 4, 5, 6 y 7).
8(b).3.8. Mezcla de ácido salicílico-ácido sulfúrico. Disolver 50 g. de ácido salicílico en dos litros de SO4 H2 concentrado.
8(b).3.9. Tiosulfato sódico en polvo. Pulverizar los cristales de S2 O3 Na2 5 H2O para que pasen por un tamiz de 1 mm. de abertura de malla.
8(b).4. Procedimiento
Colocar una muestra que contenga unos 10 mg. de N (1 g. de turba, 5 ó 10 g. de suelo y 20 g. si es muy arenoso) que haya pasado a través de un tamiz de 1 mm. de abertura de malla en un frasco seco macro-Kjeldahl y añadir 40 ml. del reactivo 8(b).3.8 y rotar el frasco para mezclar bien con el suelo. Dejar reposar la mezcla varias horas o de un día para otro, añadir 5 g. de reactivo 8(b).3.9 a través de un embudo de cuello largo que alcance la base del frasco Kjeldahl y calentar la mezcla con cuidado en la batería de digestión hasta que deje de formarse espuma. Enfriar el frasco, añadir 20 ml. de agua, 10 g. de SO4 K2, 1 g. de SO4 Cu 5 H2O y 0,1 g. de Se. Proceder a efectuar la digestión, destilación y determinación del amonio tal como se describe en 8(a).4.
8(b).5. Cálculo
Calcular el N (orgánico + nitrato) referido a porcentaje.

V = volumen en ml. de SO4 H2 0,05 N utilizado.
P = peso seco en g. de la muestra.
8(b).6. Referencia
1. Bremner, J. M.: «Methods of Soil Analysis». Part 2, p. 1.164. 1965.
9. CAPACIDAD DE CAMBIO CATIONICO
(Provisional)
9.1. Principio
El suelo se satura de sodio mediante cuatro lavados sucesivos con acetato sódico (AcONa) 1 N a pH 8,2. El exceso de sal se elimina del suelo y el sodio absorbido se desplaza con acetato amónico (AcONH4) 1 N, en cuya solución se determina el sodio.
Este método se recomienda especialmente para suelos calizos y suelos alcalinos, pero puede ser aplicado o adaptado a todos los tipos de suelos.
9.2. Material y aparatos
9.2.1. Fotómetro de llama o espectrofotómetro con accesorios para fotometría de llama.
9.2.2. Agitador mecánico de tubos de centrífuga.
9.2.3. Centrífuga y tubos de 50 ml.
9.3. Reactivos
9.3.1. Solución de acetato sódico, 1,0 N. Disolver 136 g. de acetato sódico trihidrato en agua y diluir a 1 litro. El pH de la solución debe ser, aproximadamente, 8,2.
9.3.2. Etanol al 95 por 100.
9.3.3. Solución de acetato amónico, 1 N. A 600 ó 700 ml. de agua añadir 57 ml. de ácido acético concentrado y a continuación 68 ml. de hidróxido amónico concentrado. Diluir hasta 1 litro y ajustar el pH a 7 con hidróxido amónico o ácido acético.
9.4. Procedimiento
Pesar una muestra de aproximadamente 4 g. si se trata de un suelo de textura franca o fina y una de 6 g. en el caso de un suelo de textura gruesa. Pesar las muestras con un 1 por 100 de precisión y hacer la corrección del contenido de humedad del suelo secado al aire. Colocar la muestra en un tubo de centrífuga y añadir 33 ml. de acetato sódico (reactivo 9.3.1). Agitar el tubo tapado durante cinco minutos en el agitador mecánico. Destapar y centrifugar con una fuerza centrífuga relativa, f. c. r. = 1.000 hasta que el líquido sobrenadante esté claro. Esto requiere normalmente cinco minutos. Decantar el líquido sobrenadante tan completamente como sea posible y desechar. Tratar la muestra con otras tres porciones adicionales de 33 ml. de disolución de acetato sódico 1 N, hasta un total de cuatro tratamientos. A continuación, preparar una suspensión de la muestra en 33 ml. de etanol al 95 por 100 (reactivo 9.3.2) y agitar el tubo tapado durante cinco minutos; destapar, centrifugar y decantar el líquido claro que sobrenade. Lavar la muestra de esta forma con reactivo 9.3.2 un total de tres veces. La conductividad eléctrica del líquido sobrenadante procedente del tercer lavado debe ser inferior a 40 micromhos/cm.
Desplazar el Na absorbido en la muestra con tres porciones de acetato amónico (reactivo 9.3.3) y determinar la concentración de Na en los extractos combinados después de haber diluido y mezclado en 100 ml., de acuerdo con el método 14(a).
9.5. Cálculo
Calcular la capacidad de cambio expresada en meq/100 g.

C = concentración de Na en meq/l. en el extracto.
P = peso seco en g. de la muestra.
9.6. Referencia
1. Bower, C. A.; Reiterneier, R. F., y Fireman, M.: «Exchangeable cation analysis of saline and alkali soils». Soil Ss. 73: 251-261, 1952.
10(a) CATIONES DE CAMBIO
10 (a).1. Principio
Los cationes intercambiables del suelo se desplazan mediante extracciones sucesivas con solución 1 N de acetato amónico (Ac ONH4) y se determinan en dicho extracto.
El método que se describe es aplicable a todos los suelos para la determinación de K y Na, y a suelos no calizos para la determinación de Ca y Mg.
10(a).2 Material y aparatos
10(a).2.1. Centrífuga.
10(a).2.2. Tubos de centrífuga de fondo redondo y cuello estrecho.
10(a).2.3. Vasos de 250 ml.
10(a).2.4. Agitador mecánico de tipo recíproco.
10(a).3. Reactivos
10(a).3.1. Solución de acetato amónico (Ac ONH4). 1 N. ajustada a pH 7,0; por cada litro de solución que se prepare añadir 57 ml. de ácido acético glacial a unos 600 ml. de agua, y entonces añadir 68 ml. de hidróxido amónico concentrado, de peso específico 0,90. El hidróxido debe añadirse en una vitrina de gases a través de un embudo de cuello largo de tal manera que llegue al fondo de la solución del ácido. Dejar enfriar y ajustar a pH 7,0 con ácido acético o N H4 OH, usando un pH-metro o indicador azul de bromotimol. Diluir la solución al volumen convenido, mezclarla y almacenarla en un frasco de pyrex,
10(a).3.2. Acido nítrico concentrado.
10(a).3.3. Acido clorhídrico concentrado.
10(a).3.4. Acido acético, aproximadamente 0,1 N.
10(a).4. Procedimiento
10(a).4.1. Extracción de cationes intercambiables. Pesar con precisión del 1 por 100 una muestra de suelo de aproximadamente 8 g. para los suelos de textura fina o 10 g. para los de textura gruesa. Hacer corrección del contenido de humedad de las muestras secadas al aire. Colocar el suelo en un tubo de centrífuga y añadir 33 ml. de acetato/amónico 1 N [reactivo 10(a).3.1], tapar y agitar durante cinco minutos. Quitar el tapón y centrifugar con una fuerza centrífuga relativa = 1.000 hasta que el líquido sobrenadante esté claro. Esto requiere normalmente cinco minutos. Decantar el líquido sobrenadante tan completamente como sea posible en un matraz aforado de 100 ml. Extraer con el reactivo 10(a).3.1 un total de tres veces, decantando en el matraz. Enrasar con 10(a).3.1. Transferir 50 ml. de extracto a un vaso de 250 ml. y evaporar a sequedad en placa de calentamiento o baño de agua. Lavar las paredes del vaso con una pequeña cantidad de agua y evaporar a sequedad otra vez. Añadir 1 ml. de ácido nítrico concentrado [reactivo 10(a).3.2] y 3 ml. de ácido clorhídrico concentrado [reactivo 10(a).3.3], evaporar y disolver el residuo en 20 ml. de ácido acético [reactivo 10(a).3.4]. Filtrar a través de un papel de filtro con bajo contenido en cenizas, recogiendo en un matraz de 50 ml, usando agua para lavar el vaso y el filtro. Enrasar.
Por comparación de los resultados de las determinaciones de sales solubles (método 6) y capacidad de intercambio catiónico (método 9) podrá decidirse si los cationes solubles son o no despreciables comparados con los de cambio. Cuando no lo sean se determinarán: 1.º los cationes solubles según los métodos 13(a) o 13(b) y 14, 15 y 16; 2.º el porcentaje de saturación (método 6).
10(a).4.2. Determinación de K y Na por fotometría de llama. Determinar Na y K en el extracto de acetato amónico del suelo según se describe en los métodos 14 y 15.
10(a).4.3. Determinación de Ca y Mg por complexometría. Determinar Ca y Mg en la solución acuosa de sales que han sido previamente tratadas con agua regia. El procedimiento es el descrito en el método 16.
10(a).5. Cálculo
Calcular los cationes extraídos por acetato amónico, los cationes solubles y los cationes intercambiables expresados en meq/100 g., y el potasio extraído por acetato amónico expresado en p. p. m.

C = concentración en meq/l. de catión en el extracto de acetato amónico.
P = peso en g. de la muestra de suelo referido a peso seco.

[ver métodos 13(a), 14, 15 y 16].
C' = concentración de catión en meq/l. en el extracto saturado.
S' = porcentaje de saturación.
Cationes intercambiables = (E - S) meq/100 g.
E = cationes extractables en meq/100 g.
S = cationes solubles en meq/100 g.
Potasio extraído por acetato amónico en p. p. m. = 391 · E' meq/100 g,
E' = potasio extraído por acetato amónico en meq/100 g.
10(a).6. Observaciones
En los suelos con pocas sales solubles no es necesaria la determinación correspondiente al cálculo de cationes solubles ni consiguientemente la corrección expresada por cationes intercambiables. Los cationes extraídos por acetato amónico darán directamente la cantidad de cationes intercambiables.
10(a).7. Referencia
1. Bower, C. A.; Reiterneier, R. F.; Fireman, M.: «Exchangeable cation analysis of saline and alkali soils». Soil Sci. 73: 251-261, 1952.
10(b) CATIONES DE CAMBIO: Ca y Mg
10(b).1. Principio
En los suelos calizos y dolomíticos no se puede aplicar el método 10(a) para la determinación del calcio y magnesio de cambio debido a la solubilidad de los respectivos carbonatos en la solución extractora de acetato amónico. Dichos carbonatos son muy poco solubles en una solución de acetato sódico, 1 N de pH 8,2; el error positivo debido a la disolución de dolomita se ha estimado en 0,75 meq/100 g. El uso de este factor de corrección porporciona una estimación aceptable del magnesio de cambio. El calcio de cambio se calcula por diferencia entre la capacidad de cambio catiónico y el resto de cationes cambiables, quedando, por tanto, englobados con la cifra del calcio los errores de todas las determinaciones. Esto no supone gran inconveniente ya que generalmente carece de interés práctico conocer la cantidad de calcio de cambio en suelos calizos.
10(b).2. Material y aparatos
10(b).2.1. Agitador mecánico de tubos de centrífuga.
10(b).2.2. Centrífuga.
10(b).2.3. Tubos de centrífuga de 50 ml.
10(b).3. Reactivos
10(b).3.1. Solución de acetato sódico, 1 N. Disolver 136 g de acetato sódico trihidrato en agua y diluir en 1 litro. El pH de la solución debe ser aproximadamente 8,2.
10(b).4. Procedimiento
Pesar unos 4 g. de suelo desecado al aire con un 1 por 100 de precisión y transferir a un tubo de centrífuga de 50 ml. Añadir 33 ml. de solución de acetato sódico [reactivo 10(b).3] y agitar en un agitador mecánico durante cinco minutos. Centrifugar hasta que el líquido sobrenadante esté claro y decantar el líquido en un matraz aforado de 100 ml. Repetir la extracción dos veces más. Diluir con acetato sódico y mezclar. Determinar el magnesio por complexometría (método 16).
10(b),5 Cálculo
Calcular el Mg y el Ca de cambio expresados en meq/100 g.

C = concentración en meq/l. de Mg en el extracto.
P = peso seco en g. de la muestra.
Calcio de cambio = capacidad de cambio catiónico en meq/100 g. ‒ (K + Na + Mg de cambio) en meq/100 g.
10(b).6. Observaciones
Cuando se determine la capacidad de cambio catiónico por el procedimiento indicado en el método 9, se conservará el líquido sobrenadante de los sucesivos tratamientos de la muestra con acetato sódico, determinándose en el mismo el Mg de cambio.
10(b).7. Referencia
1. Bower, C. A.: «Determination of exchangeable magnesium in soils containing dolomite». Soil Sc. Amar. Proc. 19: 40-42, 1955.
11. ACIDEZ VALORABLE (TOTAL) DE LOS SUELOS
(Provisional)
11.1. Principio
Se entiende por acidez de cambio a la parte de la acidez del suelo que puede ser reemplazada con sales neutras no tamponadas, tales como Cl Na, Cl K o Cl2 Ca. La acidez valorable es la cantidad de ácido neutralizado a un determinado pH que se toma frecuentemente a 8,2. El hidrógeno cambiable es el hidrógeno que puede ser reemplazado por sales neutras.
El método propuesto consiste en la percolación del suelo con una solución 0,5 N de Cl2 Ba y 0,055 N de trietanolamina neutralizada con ClH a pH 8. La solución que percola a través del suelo se valora con ClH hasta pH 5,1 y la cantidad de ácido que se consume se compara con el requerido en un ensayo en blanco.
De acuerdo con las anteriores definiciones en este método se determina la acidez valorable, incluyéndose conjuntamente: 1) el Al trivalente monomérico cambiable; 2) el H cambiable por cationes de sales neutras; 3) los grupos funcionales débilmente ácidos de la materia orgánica, y 4) los sesquióxidos hidrolizables asociados con minerales de arcilla o materia orgánica. Percolando una solución de sales neutras a través de un suelo se desplaza: 1) el Al trivalente monomérico cambiable, y 2) quizá algún H de la materia orgánica, aunque parece probable que el H que aparentemente a veces se encuentra procede de la hidrólisis del Al. Por tanto, ninguna de las tres expresiones, a saber, acidez valorable, acidez de cambio e hidrógeno cambiable, son sinónimas.
11.2. Material y aparatos
11.2.1. Matraces erlenmeyer de 125 y 500 ml.
11.2.2. Matraz aforado de 250 ml.
11.2.3. Filtro Büchner de pyrex, núm. 40.
11.3. Reactivos
11.3.1. Solución tampón extractora 0,5 N de Cl2 Ba 0,055 N de trietanolamina (TEA), ajustada a pH 8,0: Disolver 550 g de Cl2Ba 2H2O en agua destilada y hervida en una botella de pyrex de 10 litros. Añadir 250 ml. de trietanolamina 2 N y 36 ml. de CIH 6 N, y diluir la solución en un total de 9 litros con agua destilada y hervida (sin CO2). Mezclar bien la solución y ajustar a pH 8,00 ± 0,02 con CIH o TEA. Esta solución es 0,03 N respecto a la base libre. Impedir la disolución de CO2 en la solución, instalando un sifón y un tubo con cal sodada, conectados a la toma de aire. Para preparar esta solución extractora diluir previamente trietanolamina comercial [N (CH2 CH2 OH)3 ‒ peso especifico 1,126, aproximadamente 8 Nl para obtener una solución 2 N y normalizarla con la solución ácida patrón (reactivo 11.3.3) y el indicador (reactivo 11.3.2).
11.3.2. Solución indicadora: disolver 0,22 g. de verde de bromocresol y 0,075 g. de rojo de metilo en 96 ml. de etanol al 95 por 100 que contenga 3,5 ml. de Na OH 0,1 N.
11.3.3. Solución patrón de ácido clorhídrico, 0,2 N.
11.4. Procedimiento
Colocar 10 g. de suelo en un matraz erlenmeyer de 125 ml. y añadir 100 ml. de la solución extractora (reactivo 11.3.1), remover el contenido para mezclar bien, tapar el matraz y dejar en reposo durante una noche. Transferir el contenido a un filtro Büchner de pyrex (núm. 40), con papel de filtro Whatman núm. 32 (4,25 cm.), enjuagar el erlenmeyer con la solución extractora y continuar percolando esta solución a través del suelo hasta que se tenga un total de percolado de 225 ml. Dejar drenar cada fracción de la solución extractante antes de añadir la fracción siguiente, pero evitar que el suelo se agriete debido a secado excesivo. Transferir completamente la solución percolante a un matraz graduado de 250 ml. y enrasar con el reactivo 11.3.1. A continuación verter el contenido del matraz en un erlenmeyer de 500 ml., añadir 5 gotas de solución indicadora (reactivo 11.3.2) y valorar con Cl H (reactivo 11.3.3) hasta el viraje de color a rosa (pH 5,1). Enjuagar el matraz con la solución que se esté valorando y completar la valoración. Valorar 250 ml. de la solución extractora original hasta el mismo punto de viraje utilizando la misma cantidad de solución indicadora.
11.5 Cálculo
Calcular la acidez valorable expresada en meq/100 g. de suelo.
Acidez valorable meq/100 g. = (V ‒ V') 10 N.
V = volumen en ml. de ClH utilizado para valorar 250 ml. de la solución extractora.
V' = volumen en ml. de ClH utilizado para valorar 250 ml. de la solución extractora que ha estado en contacto con el suelo.
N = normalidad del CIH.
11.6. Observaciones.
Con algunos suelos, el color rosa del viraje desaparecerá conforme el pH aumente, debido a la lenta disolución de Al(OH)3, pero esto no hay que tenerlo en cuenta. La pequeña cantidad de aluminato disuelto en el extracto del suelo será convertido en Al(OH)3 cuando la valoración se haya completado y, por tanto, no introducirá ningún error en la determinación de la acidez total. Sin embargo, cuando la solución valorada se deje reposar, el Al(OH)3 precipitado continuará reaccionando con el ácido libre, y cualquier adición posterior de ácido para restaurar el punto de viraje introduciría un error negativo en la valoración.
11.7. Referencias
1. Peech, M.: «Methods of Soil Analysis». Part 2: 910-912. American Society of Agronomy, 1965.
2. Coleman, N. T., y Thomas. G W.: «Soil Acidity and I. ming»: 1-36. Agronomy Mon. núm. 12. American Society of Agronomy, 1967.
12. NECESIDAD DE CAL DE LOS SUELOS ACIDOS
(Provisional)
12.1. Principio
Se propone un método empírico en el que una muestra de suelo se pone en contacto con una solución tampón neutra de p-nitrofenol, basando la estimación de la necesidad de cal en el decremento de pH que produce el suelo ácido en tal solución.
12.2. Material y aparatos
12.2.1. Potenciómetro (pH-metro).
12.2.2. Electrodo de vidrio.
12.2.3. Electrodo de calomelanos.
12.3. Reactivos
12.3.1. Solución tampón de p-nitrofenol. Disolver en agua destilada 4 g. de p-nitrofenol y 18 g. de acetato cálcico anhidro, llevando a 1 litro la solución. Ajustar a pH algo inferior a 7,0 con lechada de cal concentrada obtenida a partir de óxido cálcico. Dejar en reposo durante la noche, ajustando el pH exactamente a 7,0 el día siguiente. Filtrar y almacenar en frasco de color topacio.
12.3.2. Solución de Cl K 0,1 M: Disolver 7,456 g. de Cl K en 100 ml. de agua destilada y diluir en 1 litro.
12.3.3. ClK sólido.
12.4. Procedimiento
A 10 g. de suelo tamizado y seco al aire añadir en un vaso de 100 ml. 2 g. de Cl K sólido y 25 ml. de la solución tampón (reactivo 12.3.1). Agitar varias veces durante la primera hora y ocasionalmente después, dejando los vasos cubiertos con una placa de vidrio durante la noche. A la mañana siguiente agitar de nuevo y medir el pH de la suspensión del suelo en la solución tampón. Determinar también el pH del suelo en suspensión 0,1 M de Cl K, tal como se describe en 2.4.2.
12.5. Cálculo
Calcular la necesidad de encalado expresada en Tm. de CO3 Ca por Ha., suponiendo una densidad aparente de 1,35.

pH = pH deseado en los 15 cm. superficiales de 1 Ha.
pH' = pH inicial del suelo.
pH'' = pH adquirido por la solución tampón en contacto con el suelo.
12.6. Observaciones
El método puede aplicarse siempre que el pH de la suspensión tampón no sea inferior a 5,80. El error general a este tipo de método es que la cantidad de cal para incrementar un cierto número de décimas el pH del suelo no es la misma a distintos valores de pH de suelo. En cualquier caso, la necesidad de cal que se obtenga habría que multiplicarla por un factor de corrección, generalmente comprendido entre 1,5 y 3, dependiendo de las características de la caliza, absorción de Ca por las cosechas y régimen de humedad durante el período de reacción.
12.7. Referencia
1. Guitian, F., y Muñoz, M.: «An, Edaf.» 16: 1.017-1.097, 1957.
13(a) PASTA SATURADA DEL SUELO Y SEPARACION DEL EXTRACTO
13(a).1. Principio
La conductividad eléctrica del extracto de saturación del suelo es un buen índice para estimar el efecto de las sales sobre el crecimiento de las plantas.
En el presente método se prepara la pasta saturada de suelo añadiendo agua destilada y agitando hasta que se alcanza un punto de humedad característico. La pasta se coloca en un filtro y se aplica succión para extraer una cantidad suficiente de solución para analizar.
Los datos analíticos del extracto saturado de suelo pueden ser directamente utilizados para estimar el efecto de las sales sobre el crecimiento de las plantas.
13(a).2. Material y aparatos
13(a).2.1. Recipientes para conservar extractos, tales como tubos de ensayo o frascos de 50 ml.
13(a).2.2. Recipientes de 250 ml. de capacidad como mínimo, tal como un vaso de aluminio o de vidrio grueso.
13(a).2.3. Embudo Büchner de unos 20 cm. de diámetro.
13(a).2.4. Matraz Kitasato.
13(a).2.5. Bomba o trompa de vacío.
13(a).3. Procedimiento
Preparación de la pasta. Colocar en el recipiente una cantidad de suelo variable de acuerdo con las determinaciones que se vayan a efectuar. Con un peso de 250 g. la pasta proporciona el suficiente extracto acuoso para un análisis completo. Añadir agua destilada y mezclar con la espátula hasta que se alcance la saturación. De cuando en cuando, golpear el recipiente sobre la mesa del laboratorio. En el punto de saturación la pasta es brillante a la luz, fluye lentamente cuando el recipiente se inclina y se desliza sobre la espátula sin dejar mancha excepto en los suelos muy arcillosos.
La mezcla se deja en reposo durante una hora o más y entonces debe de comprobarse la saturación y rectificarse si es necesario. Si la pasta se ha endurecido o perdido su brillantez, mezclar con más agua para conseguir la consistencia anterior. Como los suelos se embarran cuando se trabajan con el agua necesaria para llevarlos a la llamada «capacidad de campo», se debe de añadir más agua inmediatamente hasta lograr el punto de saturación. La pasta con exceso de agua se corrige añadiéndole suelo seco.
Separación del extracto. Transferir la pasta del suelo saturado al embudo Büchner vertiéndola sobre el papel de filtro colocado en el embudo y aplicar vacío. Recoger el extracto en el recipiente correspondiente (Kitasato) o bien sobre un tubo de ensayo en el interior del Kitasato y que se apoye sobre la salida del embudo. No utilizar material pyrex si se va a determinar el boro. Si la primera porción del filtrado sale turbia, debe filtrarse nuevamente o desecharse. Si se desean hacer determinaciones de carbonatos o bicarbonatos debe añadirse una solución de hexametafosfato sódico de 1.000 p. p. m., a razón de una gota por cada 25 ml. del extracto, antes de guardarlo y taparlo para evitar así la precipitación de carbonato cálcico.
Para apreciar la salinidad de los suelos, generalmente la extracción puede hacerse a los pocos minutos de haber preparado la pasta. Sin embargo, cuando el suelo contiene yeso, la conductividad del extracto saturado puede aumentar de 1 a 2 mmhos/cm. después de un período de reposo. Por tanto, en suelos yesosos, debe dejarse la pasta saturada en reposo durante varias horas antes de proceder a la extracción. En cualquier caso, si además de la conductividad eléctrica del extracto saturado se va a analizar la composición química de las sales disueltas, la pasta saturada debe de dejarse en reposo de cuatro a dieciséis horas antes de extraer la solución.
13(a).4. Observaciones
Para determinaciones en serie, en suelos de textura uniforme, se puede ganar tiempo determinando la humedad de saturación de una muestra representativa según se describe en el método 40. Las otras muestras se saturarán añadiendo el suficiente volumen de agua a pesos conocidos de suelos.
Las pastas saturadas de suelos turbosos deben de dejarse en reposo durante toda una noche para comprobar la saturación.
En los suelos de textura fina para reducir la tendencia a adquirir consistencia de barro, y obtener un punto final más definido, el agua debe de añadirse a estos suelos con una mínima operación de, movimiento, especialmente al princpio.
En los suelos de textura gruesa la pasta puede ser preparada en la misma forma que para los de textura fina. No obstante, se recomienda hacer el extracto a un diferente contenido de humedad para la determinación de salinidad [método 13(b)].
13(a).5. Referencia
1. United States Salinity Laboratory Staff: «Diagnosis and Improvement of Saline and Alkali Soils», 1954.
13(b) EXTRACTO DE SATURACION DOBLE PARA SUELOS DE TEXTURA GRUESA
13(b).1. Principio
En el siguiente procedimiento se prepara una pasta con doble humedad que la existente en el punto de saturación. La conductividad del extracto de saturación doble hay que multiplicarla por dos antes de usar la escala patrón de evaluación de salinidad del suelo 13(a).4.
13(b).2. Material y aparatos
13(b).2.1. Lata de un diámetro de 10 a 12 cm.
13(b).2.2. Cesto de tela metálica galvanizada, con aberturas de 6 mm2 aproximadamente y que se ajuste a la lata.
13(b).3. Procedimiento
Colocar el cesto en la lata y añadir la muestra de suelo al cesto hasta formar una capa de 2 ó 3 mm. de espesor. Nivelar el suelo y añadir con pipeta 2 ml. de agua, gota a gota, en áreas contiguas del suelo. Cubrir el cesto y dejar reposar durante quince minutos. Tamizar suavemente el suelo seco a través de la tela metálica del cesto y pesar las bolitas de suelo húmedo retenidas.
Calcular la humedad como sigue:

H = contenido en humedad.
P = peso en g. referido a materia húmeda.
Pesar 250 g. de suelo seco al aire y añadir suficiente agua hasta cuatro veces el contenido en humedad H. Después de un periodo de reposo, proceder como en 13(a).3.2.
13(b).4. Observaciones
Todos los resultados de determinaciones que se hagan sobre el extracto de saturación doble habrán de ser multiplicados por dos antes de interpretar los valores en la escala patrón de salinidad.
13(b).5. Referencia
1. United States Salinity Laboratory Staff: «Diagnosis and Improvement of Saline and Alkali Soils», p. 88, 1954.
14(a) SODIO POR FOTOMETRIA DE LLAMA
14(a).1. Principio
Excitación del Na de soluciones pulverizadas sobre una llama, y medida de la radiación emitida, utilizando Li como patrón interno para compensar las variaciones en las condiciones de funcionamiento del fotómetro de llama y reducir las interferencias.
14(a).2. Material y aparatos
14(a).2.1. Matraces aforados de 50 ml.
14(a).2.2. Fotómetro de llama.
14(a).3. Reactivos
14(a).3.1. Solución de acetato amónico, aproximadamente 1 N. Añadir 57 ml. de ácido acético concentrado a unos 600 ml. de agua y luego 68 ml. de hidróxido amónico concentrado. Diluir en un volumen de 1 litro y ajustar a pH 7,0, añadiendo hidróxido amónico a ácido acético.
14(a).3.2. Solución de cloruro sódico 0,04 N. Disolver 2,338 gramos de cloruro sódico desecado en agua y diluir exactamente en 1 litro.
14(a).3.3. Solución de cloruro sódico 0,04 N en acetato amónico 1 N. Disolver 2,338 g. de cloruro sódico desecado en reactivo 14(a).3.1 hasta completar 1 litro.
14(a).3.4. Solución de cloruro de litio 0,05 N. Disolver 2,12 gramos de cloruro de litio desecado en agua y diluir exactamente en 1 litro.
14(a).4. Procedimiento
Preparar una serie de patrones de cloruro sódico usando los reactivos 14(a).3 (2 y 4), añadiendo a cada uno la misma alícuota de la solución 14(a).3.4. Preparar una serie similar de patrones de cloruro sódico con los reactivos 14(a).3.3 y 14(a).3.4, usando, el reactivo 14(a).3.1 en vez de agua para diluir. Las concentraciones de cloruro sódico que se recomiendan son 0; 0,2; 0,6; 0,8; 1; 2; 3 y 4 meq/l. La concentración óptima de cloruro de litio varía con el tipo de fonómetro de llama, pero generalmente es de 5 a 10 meq/l. Los patrones de cloruro sódico en agua se usan para análisis del Na en aguas y en extractos acuosos de suelos. Los patrones de cloruro sódico en acetato amónico se usan para análisis del Na en el extracto de acetato amónico del suelo. Calibrar el fotómetro para un intervalo de concentraciones entre 0 y 1 meq/l. de Na, usando las seis primeras soluciones patrones. Usar la primera y las cuatro últimas soluciones patrones cuando se vaya a trabajar en el intervalo de 0 a 4 meq/l. de Na.
Medir con pipeta una alícuota de la solución que se va a analizar en un matraz aforado de 50 ml., la alícuota debe de contener menos de 0,2 meq. de Na. Añadir el volumen apropiado del reactivo 14(a).3.4 de modo que cuando se diluya a un volumen de 50 ml. la concentración de litio sea exactamente igual a la de las soluciones patrón de cloruro sódico. Enrasar, añadiendo agua o reactivo 14(a).3.1, si van a analizarse extractos de acetato amónico. Mezclar y determinar la concentración de Na con el fotómetro de llama usando la curva de calibración apropiada.
14(a).5. Cálculo
Calcular el contenido en Na en agua o extracto del suelo expresado en meq/l.

C = concentración de Na en meq/l. hallada en la curva de calibración.
V = volumen en ml. de la alícuota de la solución a analizar.
14(a).6. Observaciones
Usando el patrón interno de cloruro de litio tal como se prescribe en este método se atenúan los efectos de las variaciones de presión del combustible y del oxígeno, los cambios de viscosidad, moléculas orgánicas y de sales y ácidos inorgánicos. No es necesario que la concentración de litio en el patrón interno sea exacta pero sí que sea la misma concentración y de la misma sal tanto en los patrones como en las soluciones problemas. Ver manual de instrucciones del fotómetro que se utilice.
14(a).7. Referencia
1. United States Salinity Laboratory Staff: «Diagnosis and Improvement of Saline and Alkali Soils, 96, 1954.
14(b) SODIO POR PRECIPITACION COMO ACETATO DE URANILO, CINC Y SODIO
14(b).1. Principio
Determinación gravimétrica del sodio precipitado con acetato de uranilo y cinc.
14(b).2. Material y aparatos
14(b).2.1. Centrífuga.
14(b).2.2. Tubos cónicos de centrífuga de 12 ml.
14(b).3. Reactivos
14(b).3.1. Acetato de uranilo y cinc: Pesar 300 g. de acetato de uranilo dihidratado, 900 g. de acetato de cinc dihidratado y 10 g. de cloruro de sodio y llevar a un matraz grande. Añadir 82 ml. de ácido acético glacial y 2.618 ml. de agua. Agitar hasta que las sales se disuelvan, dejando sólo una pequeña cantidad de acetato de uranilo, cinc y sodio. Filtrar antes de usar.
14(b).3.2. Acido acético-etanol: Mezclar 150 ml. de ácido acético glacial con 850 ml. de etanol al 95 por 100. Agitar con un exceso de cristales de acetato de uranilo, cinc y sodio. Filtrar antes de usar. Los cristales de acetato de uranilo, cinc y sodio pueden prepararse como sigue: Añadir 125 ml. de acetato de uranilo y cinc [14(b).3.1] a 5 ml. de solución de cloruro sódico al 2 por 100, agitar y después de quince minutos recoger el precipitado en un crisol de porcelana de fondo poroso. Lavar varias veces con ácido acético glacial, luego varias veces con éter y, finalmente, desecar en un desecador.
14(b).3.3. Eter anhidro.
14(b).4. Procedimiento
Pipetar una alícuota que contenga de 0,003 a 0,07 meq de sodio en un tubo de centrífuga cónico de 12 ml. Evaporar en baño de agua hasta 0,5 ml. Enfriar, añadir 8 ml. de acetato de uranilo y cinc [14(b).3.1], y mezclar agitando con un alambre de aluminio doblado en un lazo. Dejar en reposo una hora. Centrifugar a f. c. r. = 1.000 durante diez minutos. Decantar y drenar sobre papel de filtro durante diez minutos. Secar la boca del tubo con un paño limpio o con papel de filtro sin hilachas. Suspender el precipitado y lavar las paredes del tubo, utilizando 5 ml. de ácido acético-etanol [14 (b).3.2] vertidos con una pipeta equipada de una pera de goma. Centrifugar durante diez minutos, decantar y drenar durante un minuto. Secar la boca del tubo. Lavar con 5 ml. de éter anhidro [14(b).3.3], y centrifugar sólo durante cinco minutos. Decantar cuidadosamente sin drenar. Repetir el lavado y la centrifugación otra vez. Limpiar el exterior del tubo con gamuza, desecar durante una hora o más a 60° C, enfriar en un desecador y pesar.
Añadir 10 ml. de agua, agitar con el alambre hasta que se disuelva el precipitado de sodio, centrifugar durante cinco minutos, decantar cuidadosamente y drenar durante cinco minutos sobre papel de filtro. Suspender el precipitado insoluble y lavar las paredes del tubo con 5 ml. de ácido acético-etanol [14(b).3.2] vertidos con pipeta. Centrifugar durante cinco minutos y decantar. Lavar con 5 ml. de éter anhidro, centrifugar una hora a 60° C, enfriar en un desecador y pesar. La diferencia entré los dos pesos es el peso del precipitado de sodio.
14(b).5. Cálculo

P = peso en g. del sodio precipitado.
V = volumen en ml. de la alícuota.
14(b).6. Referencias
1. Reiterneier, R. F.: «Semimicroanalysis of saline soils Solutions». Indus. and Engin. Chem. Analyt. 15: 393-402, 1943.
2. United States Salinity Laboratory Staff: «Diagnosis and Improvement of Saline and Alkali Soils». p. 97, 1954.
15(a) POTASIO POR FOTOMETRIA DE LLAMA
15(a).1. Principio
Como en 14(a).1.
15(a).2. Material y aparatos
15(a).2.1. Matraces aforados de 50 ml.
15(a).2.2. Fotómetro de llama.
15(a).3. Reactivos
15(a).3.1. Solución de acetato amónico, aproximadamente 1 N. Añadir 57 ml. de ácido acético concentrado a unos 600 ml. de agua y luego unos 68 ml. de hidróxido amónico concentrado. Diluir en un volumen de 1 litro y ajustar pH 7, añadiendo más hidróxido amónico o ácido acético.
15(a).3.2. Solución de cloruro potásico, 0,02 N. Disolver en agua 1,491 g. de cloruro potásico desecado y diluir en un volumen de 1 litro.
15(a).3.3. Solución de cloruro potásico 0,02 N en acetato amónico 1 N. Disolver 1,491 g. de cloruro potásico desecado en reactivo 15(a).3.1 y completar hasta 1 litro con dicho reactivo.
15(a).3.4. Solución de cloruro de litio, 0,05 N. Disolver en agua 2,12 g. de cloruro de litio desecado y diluir en 1 litro.
15(a).4. Procedimiento
Preparar una serie de patrones de cloruro potásico usando los reactivos 15(a).3.2 y 15(a).3.4, añadiendo a cada uno la misma alícuota de la solución. Preparar una serie similar de patrones de cloruro potásico con los reactivos 15(a).3.3 y 15(a).3.4 usando el reactivo 15(a).3.1. en vez de agua para diluir. Las concentraciones de cloruro potásico que se recomiendan son: 0; 0,1; 0,2; 0,3; 0,4; 0,5; 1; 1,5 y 2 meq/l. Los patrones de cloruro potásico en agua se usan para análisis de aguas y extractos acuosos de suelos. Los patrones de cloruro potásico en acetato amónico se usan para el análisis del K en el extracto de acetato amónico del suelo. Calibrar el fotómetro para él intervalo de concentración de 0 a 0,5 meq/l. usando los seis primeros patrones de la serie. Usar el primer patrón y los cuatro últimos para calibrar el fotómetro en el intervalo de 0 a 2 meq/l. de potasio.
Medir con pipeta una alícuota que contenga menos de 0,1 meq. de potasio de la solución problema y colocarla en un matraz aforado de 50 ml. Añadir el volumen apropiado del reactivo 15(a).3.4, de modo que cuando se diluya a un volumen de 50 ml. la concentración de litio en la solución problema sea exactamente igual a la de las soluciones, patrón de cloruro potásico. Enrasar añadiendo agua o reactivo 15(a).3.1 en el caso de que se vayan a analizar extractos de acetato amónico. Mezclar y determinar la concentración de potasio con el fotómetro de llama, usando la curva de calibración apropiada.
15(a).5. Cálculo
Calcular el contenido de K en agua o extracto del suelo expresado en meq/l.

C = concentración en meq/l. de K hallada en la curva de calibración.
V = volumen en ml, de la alícuota de la solución a analizar.
15(a).6. Observaciones
Usando el patrón interno de cloruro de litio tal como se prescribe en este método se atenúan los efectos de las variaciones de presión del combustible y del oxígeno, los cambios de viscosidad, moléculas orgánicas y de sales y ácidos inorgánicos. No es necesario que la concentración de litio en el patrón interno sea exacta, pero si que sea la misma concentración y de la misma sal tanto en los patrones como en las soluciones problemas.
15(a).7. Referencia
1. United States Salinity Laboratory Staff: «Diagnosis and Improyement of Saline and Alkali Soils», 97, 1954,
15(b) POTASIO POR PRECIPITACION COMO DIPICRILAMINATO DE POTASIO
15(b).1. Principio
Determinación colorimétrica del potasio precipitado con dipicrilaminato de litio.
15(b).2. Material y aparatos.
15(b).2.1. Colorímetro fotoeléctrico.
15(b).2.2. Centrífuga.
15(b).2.3. Tubos cónicos de centrífuga de 12 ml.
15(b).3. Reactivos.
15(b).3.1. Solución de dipicrilaminato de litio. Disolver 1,65 gramos de carbonato de litio en 250 ml. de agua. Calentar a 50° C y luego añadir 9 g. de dipicrilamina. Después de que la dipicrilamina se haya disuelto, filtrar y diluir 200 ml. de esta solución a 1 litro. Para la porción restante, de aproximadamente 50 ml., añadir 0,25 g. de cloruro potásico. Separar y lavar el precipitado de dipicrilaminato de potasio resultante con unos pocos mililitros de agua mediante una centrífuga. Añadir la sal de potasio a la solución caliente de dipicrilaminato de litio y agitar durante treinta minutos. Filtrar la solución antes de usar.
15(b).3.2. Cloruro potásico, 0,01 N. Disolver 0,7456 g. de cloruro potásico seco en agua y diluir, a 1 litro exactamente.
15(b).3.3. Fenolftaleína al 1 por 100 en etanol del 60 por 100.
15(b).3.4. Hidróxido sódico, aproximadamente 1 N. Disolver 40 g. de hidróxido sódico en agua y diluir a 1 litro.
15(b).4. Procedimiento
Pipetar una alícuota que contenga 0,005-0,035 meq. de potasio en un tubo cónico de centrífuga de 12 ml. Añadir una gota de fenolftaleína [15(b).3.3] y luego hidróxido sódico [15(b).3.4] hasta coloración rosa. Evaporar a sequedad. Esto asegura la eliminación del amonio. Enfriar y luego añadir exactamente 2 ml. de solución de dipicrilaminato de litio [15(b).3.1]. Triturar el residuo salino en el fondo del tubo con una varilla de vidrio y dejar precipitar durante una hora. Centrifugar el tubo durante un minuto a f. c. r. = 1.000. Tomar una alícuota de 0,2 ml. del líquido sobrenadante con una pipeta de sangre y diluir hasta un volumen de 50 ml. Comparar la transmisión en una cubeta óptica a través de un filtro de 510 mμ con la del agua en una cubeta igual. Preparar una curva de calibración para cada grupo de muestras sometiendo una serie de 0,5; 1; 1,5; 2; 2,5; 3; 3,5 ml. de cloruro potásico [15(b).3.2] a las mismas operaciones anteriormente descritas. La cantidad de potasio en la muestra se encuentra interpolando en esta curva. Representada a escala lineal la curva debe tener una forma ligeramente de S. La temperatura de preparación de la curva de calibración no debe diferir en más de 2° C de la temperatura a la que se hacen las determinaciones problema.
15(b).5. Cálculo

M = miliequivalentes de potasio en la alícuota determinados por interpolación en la curva de calibración.
V = volumen en ml. de la alícuota.
15(b).6. Referencias
1. Williams, W. O.: «Rapid determination of potassium with dipicrilamina». Amer. Soc. Hort. Sci. Proc. 39: 47-50, 1941.
2. United States Salinity Laboratory Staff: «Diagnosis and Improvement of Saline and Alkali Soils», p. 98, 1954.
16. CALCIO + MAGNESIO Y CALCIO POR COMPLEXOMETRIA
(provisional)
16.1. Principio
El método propuesto es el de Diehl et al. (1950), tal como es descrito por el U. S. Salinity Laboratory Staff (1954). Es aplicable a los extractos acuosos de saturación de suelo y a otros extractos no acuosos, con las precauciones que se señalan.
16.2. Material y aparatos
16.2.1. Microbureta de 10 ml.
16.2.2. Erlenmeyer de 125 ml.
16.3. Reactivos
16.3.1. Solución tampón de cloruró amónico e hidróxido amónico. Disolver 67,5 g. de cloruro amónico en 570 ml. de hidróxido amónico concentrado y completar hasta un litro.
16.3.2. Hidróxido sódico aproximadamente 4 N. Disolver 160 g. de hidróxido sódico en un litro de agua
16.3.3. Solución patrón de CI2 Ca, 0,01 N. Disolver 0,500 g. de carbonato cálcico puro (cristales de calcita) en 10 ml. de ácido clorhídrico aproximadamente 3 N (1 + 3) y diluir con agua hasta un litro.
16.3.4. Indicador de eriocromo negro T. Disolver 0,5 g. de eriocromo negro T y 4,5 g. de hidrocloruro de hidroxilamina en 100 ml. de etanol del 95 por 100. Este indicador se puede adquirir bajo diversas denominaciones comerciales.
16.3.5. Indicador de purpurato amónico (murexida). Mezclar bien 0,5 g. de purpurato amónico con 100 g. de sulfato potásico pulverizado.
16.3.6. Solución de versenato, aproximadamente 0,01 N (EDTA, saldisódica y de dihidrógeno del ácido etilenamintetracético). Disolver 2 g. de la sal y 0,05 g. de cloruro magnésico hexahidrato en agua y diluir en un litro. Calcular el título exacto de esta solución, valorándola con el reactivo 16.3.3., según el procedimiento que se detalla más adelante. La solución debe de ser valorada dos veces usando cada vez el indicador 16.3.4 y 16.3.5, ya que la normalidad que se obtiene con murexida 16.3.5 es del 3 al 5 por 100 más alta que con el eriocromo negro 16.3.4. Conservar la solución en frasco de polietileno en los cuales la normalidad no varía.
16.4. Procedimiento
16.4.1. Pretratamiento de los extractos del suelo. En los extractos acuosos de saturación del suelo se podrá, en general, proceder directamente a la valoración de Ca y Mg. Sin embargo, como el que se prepara para la obtención de cationes cambiables [métodos 10.(a) y 10(b)], es necesario eliminar el acetato amónico y la materia orgánica dispersada antes de valorar con versenato, tal como se describe en 10(a).4.1.
16.4.2. Determinación del calcio. Tomar una alícuota de 5 a 25 ml. que contenga como máximo 0,1 meq. de Ca en un erlenmeyer de 125 ml. Diluir hasta 25 ml., agregar 0,25 ml. (5 gotas) del reactivo 16.3.2 y unos 50 mg. del reactivo 16.3.5 y valorar con solución 16.3.6 usando una microbureta de 10 ml. El cambio de color sobre fondo blanco es de rojo anaranjado a morado. Cuando se aproxime el punto de viraje, el versenato debe de añadirse poco a poco (una gota cada cinco o diez segundos), ya que el cambio de color no es instantáneo.
Un ensayo en blanco conteniendo los reactivos 16.3.2 y 16.3.5 y una o dos gotas de reactivo 16.3.6 ayuda a distinguir el punto de viraje. Si se sobrepasa el punto de viraje se puede valorar por retroceso con el reactivo 16.3.3.
16.4.3. Determinación de calcio + magnesio. Tomar una alícuota de 5 a 25 ml. que contenga como máximo 0,1 meq. de Ca + Mg en un erlenmeyer de 125 ml. Diluir hasta 25 ml., aproximadamente, y agregar 0,50 ml. (10 gotas) del reactivo 16.3.1 y tres o cuatro gotas de reactivo 16.3.4. Valorar con el reactivo 16.3.6 usando una microbureta de 10 ml. El cambio de color es de rojo vino a azul o verde. Ningún matiz de rojo de vino debe permanecer en el punto de viraje.
16.5. Cálculo
Calcular el contenido de Ca o Ca + Mg en agua o extracto del suelo en meq/l.

V = volumen en ml. de la solución de versenato.
N = normalidad de la solución de versenato.
V' = volumen en ml. de la alícuota.
16.6. Observaciones
Cuando hierro, aluminio y manganeso están presentes en concentraciones mayores de 20 p. p. m. y de varias décimas de p. p. m. para el Cu, interfieren con la determinación con indicador de eriocromo negro. Normalmente, las concentraciones de estos metales en los extractos acuosos o de acetato amónico de los suelos de las regiones áridas están por debajo de esos límites. Si se produjesen interferencias pueden eliminarse por el procedimiento descrito por Cheng y Bray (1951).
16.7. Referencias
1. Cheng, K.L., y Bray, R. H.: «Determination of calcium and magnesium in soil and plant material». Soil Sc. 72: 449-458, 1951.
2. Diehl, H.; Goetz, C. A., y Hach, C. C.: «The versenate titration for total hardness». Amer. Water Works Assoc. Jour 452: 40-48, 1950.
3. United States Salinity Laboratory Staff: «Diagnosis and Improvement of Salino and Alkali Soils», pp. 94-95, 1954.
17. SULFATOS
(Provisional)
17.1. Principio
El sulfato se precipita como sulfato cálcico en presencia de acetona. El sulfato cálcico es posteriormente separado por centrifugación y redisuelto en agua. En la solución acuosa se determina la cantidad de sulfato por valoración del calcio con versenato. El método es rápido y suficientemente preciso para análisis de rutina de aguas y extractos acuosos de suelos, pero no se recomienda para suelos con bajo contenido en sulfatos (<20 p.p. m.).
El método más preciso para la determinación de sulfatos es la determinación gravimétrica por precipitación de SO4 Ba. Este método es laborioso y dudosamente recomendable para análisis rutinarios. En este tipo de análisis está frecuentemente justificada la estimación de la concentración de sulfatos por diferencia, en meq/l., entre la suma de cationes Ca2+ + Mg2+ + K+ + Na+, y la suma de los aniones determinados Co32‒ + CO3 H‒ + Cl‒.
17.2. Material y aparatos
17.2.1. Centrífuga.
17.2.2. Tubos de centrífuga de fondo cónico de 40 ó 50 ml.
17.2.3. Microbureta de 10 ml.
17.2.4. Erlenmeyer de 125 ml.
17.3. Reactivos
17.3. (1, 2, 3, 4 y 5). Corno 16.3.2, 3, 4, 5 y 6.
17.3.6. Solución de rojo de metilo, al 0,1 por 100 en alcohol.
17.3.7. Acido acético: Añadir 1 ml. de ácido acético glacial a 100 ml. de agua.
17.3.8. Acetona,
17.3.9. Solución de cloruro cálcico 1 N. Disolver 11 g de Cl2 Ca o 14,7 g. de Cl2 Ca 2 H2O y diluir hasta 100 ml.
17.4. Procedimiento
Transferir una alícuota que contenga 0,01 a 0,10 meq. de sulfato a un tubo de centrífuga de fondo cónico. Diluir o concentrar a 10 ml. Añadir una gota de rojo de metilo y ácido acético (17.3.7) gota a gota hasta que aparezca un color rosado permanente. Mezclar bien el contenido después de la adición de cada gota de ácido. Añadir 1 ml. de Cl2 Ca 1 N y mezclar. Añadir 20 ml. de acetona y mezclar con una varilla. Dejar reposar durante diez minutos para que el precitado flocule, centrifugar durante cinco minutos y decantar el líquido sobrenadante. Invertir el tubo y drenar sobre papel de filtro durante cinco minutos. Añadir 10 ml. de acetona de un frasco lavador y disgregar el precipitado enjuagando las paredes del tubo. Centrifugar y drenar de nuevo. Añadir 10 ml. de agua para disolver el precipitado. Valorar el Ca con EDTA tal como se describe en el método 13.
17.5. Cálculo
Calcular el contenido en sulfatos expresados en meq/l.

V = volumen en ml. de EDTA utilizados en la valoración.
N = normalidad de EDTA.
V' = volumen en ml. de alícuota valorado.
17.6. Observaciones
Una variante simple del método propuesto consiste en medir la conductividad eléctrica de la solución acuosa de sulfato cálcico tal como se describe en el método 22 en vez de determinar el calcio por complexometria.
17.7. Referencia
1. Branson, R., y Sharpless, R.: «Calcium Sulfate method for the determination of sulfate» (sin publicar). Dept. of Soils and Plant Nutrition. University of California. Riverside, 1957.
18. CARBONATOS Y BICARBONATOS EN EXTRACTOS DE SUELO
18.1. Principio
18.2. Material y aparatos
18.2.1. Microbureta de 10 ml.
18.3. Reactivos
18.3.1. Fenolftaleína al 1 por 100, en etanol al 60 por 100.
18.3.2. Anaranjado de metilo, 0,01 por 100 en agua.
18.3.3. Acido sulfúrico, aproximadamente 0,01 N, de titulo exacto conocido.
10.4. Procedimiento
Transferir una alícuota de la solución problema que contenga 0,005 a 0,04 meq/l. de cloruro en un crisol de porcelana de boca ancha y 15 ml. de capacidad. Se especifica la cantidad de cloruros porque la misma muestra se usa para determinar posteriormente los cloruros [método 19 (a) o 19(b)]. Añadir una gota de fenolftaleína. Si la solución toma color rosa, añadir el ácido sulfúrico usando una microbureta de 10 ml. gota a gota y a intervalos de cinco segundos, hasta que desaparezca el color. Añadir dos gotas de anaranjado de metilo y valorar con ácido hasta que aparezca el primer color anaranjado. Conservar la muestra si se van a analizar cloruros.
Para corregir por la posible alcalinidad de los indicadores, valorar una muestra en blanco de agua destilada y hervida, a la que se añaden las mismas cantidades de indicadores que a la solución problema. Restar las lecturas obtenidas si no son despreciables de las correspondientes al problema. La luz debe ser adecuada para apreciar los cambios de colores. Conviene tener a la vista soluciones de colores representativos de los puntos de viraje de las valoraciones.
18.5. Cálculo
Calcular el contenido en carbonatos y bicarbonatos expresados en meq/l.


N = normalidad de SO4H2.
V = volumen en ml. de SO4H2 utilizado para desaparición de color rosa.
V' = volumen en ml. de SO4H2 utilizado para aparición de color anaranjado primitivo
V" = volumen en ml. de la alícuota de solución problema.
18.6. Referencia
1. United States Salinity Laboratory Staff: Diagnosis and Improvement of Saline and Alkali Soils»: p. 98, 1954.
19(a) CLORUROS
(Indicador)
19(a).1. Principio
Los procedimientos clásicos para determinación de cloruros se basan en la formación de una sal de plata relativamente insoluble.
En la práctica de análisis agrícolas se llevan a cabo determinaciones volumétricas más que gravimétricas. El punto de viraje de la valoración de cloruros con nitrato de plata puede ser detectado de diversas maneras, tal como por la aparición de un precipitado rojo de Cr O4 Ag2 (valoración de Mohr), o midiendo el potencial que se desarrolla en la solución mediante una combinación apropiada de electrodos (Kolthoff y Kuroda, 1951). Se proponen ambas variantes del método.
19(a).2. Material y aparatos
19(a).2.1. Microbureta de 10 ml.
19(a).3. Reactivos
19(a).3.1. Solución al 5 por 100 de cromato potásico. Disolver 5 g. de cromato potásico en 50 ml. de agua y añadir nitrato de plata 1 N gota a gota hasta que se forme un precipitado rojo claro permanente. Filtrar y diluir hasta 100 ml.
19(a).3.2. Nitrato de plata, 0,005 N. Disolver 0,8495 g. de secado en estufa de nitrato de plata en agua y diluir exactamente en un litro. Almacenar la solución en un frasco color topacio para protegerla dé la luz.
19(a).4. Procedimiento
Tomar una alícuota de la solución problema y valorar con ácido sulfúrico hasta el punto de viraje con naranja de metilo (método 18). Se usará una solución patrón del ácido en caso de que interese determinar carbonatos y bicarbonatos.
Añadir cuatro gotas de reactivo 19(a).3.1 a la muestra guardada después de la determinación de carbonatos bicarbonatos. Valorar, agitando al mismo tiempo bajo luz brillante, con el reactivo 19(a).3.2, usando una microbureta de 10 ml., hasta que aparezca el primer color rojo (pardo rojizo) permanente.
Las correcciones del ensayo en blanco varían con el volumen de la muestra en el punto de viraje, y generalmente aumenta regularmente entre 0,03 a 0,20 ml., según aumenta el volumen de 2 a 12 ml.
19(a).5. Cálculo
Calcular la concentración de cloruros en meq/l.

V = volumen en ml. de NO3Ag utilizados en la valoración de la solución del problema.
V' = volumen en ml. de NO3Ag utilizados en la valoración en blanco.
V'' = volumen en ml. de alícuota de solución problema.
19(a).6. Observaciones
Los ácidos minerales que disuelven los cromatos de plata deben de estar ausentes. Por tanto, la cantidad de ácido añadida debe ser la estrictamente necesaria para hacer la solución débilmente ácida, evitándose la adición en exceso. Los ioduros, bromuros y bicarbonatos forman precipitados con nitrato de plata y deben estar ausentes de la muestra. Generalmente, los fosfatos no interfieren pero también deben de evitarse cantidades altas de fosfato. Los materiales orgánicos también deben de eliminarse por su tendencia a reducir el nitrato de plata en soluciones neutras. El hierro normalmente reacciona con el cromato potásico produciendo un cromato insoluble, pero la cantidad de hierro que generalmente se encuentra en las muestras no interfiere. En caso de que se crea que la cantidad de hierro es alta, añadir unas cuantas gotas más de indicador para asegurar el exceso de cromato.
19(a).7. Referencias
1. Chapman, H. D., y Pratt, P. F.: «Methods of Analysis for Soils, Plants and Waters», 98-100, 1961.
2. Reitmeier, R. F.: «Semimicroanalysis of saline soil Solutions». Indus. and Engin. Chem. Analyt. Ed. 15: 393-402, 1943.
3. Stout, P. R., y Johnson, C. M.: «Methods of Soil Analysis». Part 2: 1.125-1.126. American Society of Agronomy, 1965.
19(b) CLORUROS
(Potenciometría)
19(b).1. Principio
Ver 19(a).1. El método descrito es aplicable al extracto de saturación del suelo (o a otro extracto de diferente proporción suelo : agua) y a una suspensión de suelo : agua en el caso de que se quiera ahorrar el trabajo de separación del extracto.
19(b).2. Material y aparatos
19(b).2.1. Potenciómetro (pH-metro).
19(b).2.2. Electrodo de vidrio.
19(b).2.3. Electrodo de plata.—Cloruro de plata (Ag-Cl Ag).
19(b).2.4. Microbureta de 5 ó 10 ml.
19(b).2.5. Agitador magnético.
19(b).3. Reactivos
19(b).3.1. Suspensión de cloruro de plata (Cl Ag). Preparar un precipitado de Cl Ag mezclando soluciones 0,1 N de Cl Na y NO3 Ag; 500 ml. de cada solución serán suficientes para precipitar Cl Ag para muchas determinaciones. Debe de usarse un ligero exceso de cloruro o de nitrato para provocar un buen precipitado. Lavar bien el precipitado con agua destilada y transferirlo a un frasco topacio. Renovar la solución sobrenadante diariamente durante diez días, reemplazándola con agua destilada para remover las trazas de los excesos de iones plata o cloruro. Finalmente, completar el volumen con agua hasta un litro.
19(b).3.2. Solución de nitrato de plata (NO3 Ag), 0,1 N. Disolver 8,5 g. de NO3 Ag en agua destilada, diluir la solución en un volumen de 500 ml. y almacenar en un frasco topacio.
19(b).3.3. Solución de nitrato de plata (NO3 Ag), 0,01 N. Diluir 10,00 ml. de reactivo 19(b).3.2 en 100 ml. con agua destilada. Comprobar antes de usarla su normalidad valorándola potenciométricamente con la solución de Cl K [reactivo 19(b).3.4], según se describe más adelante.
19(b).3.4. Solución patrón de cloruro potásico (Cl K), 0,01 N. Disolver 0,746 g. de Cl K puro y desecado en estufa y diluir hasta un litro. Esta solución es 0,01 N con respecto al cloruro y se usa para valorar la solución de NO3 Ag.
19(b).3.5. Solución soporte de electrolito. Disolver 101 g. de nitrato potásico (NO3 K) recristalizado (calidad reactivo) en agua, añadir 62 ml. de ácido - nítrico concentrado (NO3 H) y diluir la solución en un litro.
19(b).4. Procedimiento
Transferir una alícuota de la solución problema, preferentemente que contenga menos de 0,1 meq. de cloruros, al vaso de valoración. Diluir en agua destilada hasta un total de 25 ml., aproximadamente. Añadir 2,5 ml. de la solución de electrolito.
Preparar la solución de referencia añadiendo a 25 ml. de agua destilada 2,5 ml. de la solución soporte de electrolito y dos gotas de la suspensión de Cl Ag. Sumergir el juego de electrodos de referencia, agitando ligeramente; cerrar el circuito del pH-metro y anotar el potencial observado en milivoltios. Reemplazar la solución de referencia por la solución problema que contenga la solución soporte de electrolito. Valorar agitando suavemente con NO3 Ag 0,01 N hasta que el potencial que indique la escala del potenciómetro o pH-metro sea el mismo que el anotado para la solución de referencia. Anotar el volumen de solución de NO3 Ag requerido.
19(b).5. Cálculo
Calcular la concentración de Cl‒ en la solución problema, expresada en meq/l.

V = volumen en ml. de la solución de NO3 Ag.
N = normalidad de la solución de NO3 Ag.
V' = volumen en ml. de la solución problema.
19(b).6. Observaciones
19(b).6.1. Los vasos de valoración no deben de exponerse a la luz solar directa durante la volaración, lo que podría provocar un cambio del potencial final. La agitación excesivamente rápida también puede cambiar el potencial final añadiendo una componente de potencial de corriente.
19(b).6.2. El uso de la solución soporte de electrolito de alta fuerza iónica, dispensa de conocer el verdadero potencial de equivalencia al mismo tiempo de ahorrar las correcciones necesarias por diferencias de fuerza iónica de la solución problema o clase de electrolitos disueltos. El «potencial aparente de equivalencia» que se obtiene es prácticamente independiente de las valoraciones normales debidas al diferente contenido de sales solubles de la solución problema.
19(b).6.3. El procedimiento descrito no diferencia entre los iones cloruros y bromuros.
19(b).7. Referencias
1. Kolthoff, I. M., y Kuroda, P. K.: «Determination of traces of chloride». Anal. Chem. 23: 1.304-1.306, 1951.
2. Stout, P. R., y Johnson, C. M.: «Methods of Soil Analysis». Part 2, 1.127-1.129, American Society of Agronomy, 1965.
20. NITRATOS
(Provisional)
20.1. Principio
Los nitratos se extraen del suelo con solución saturada de SO4 Ca. 2H2O y se determinan en el filtrado por el método del ácido fenoldisulfónico. Para ello, se evapora una alícuota del extracto a sequedad después de añadir CO3 Ca para impedir la pérdida de nitrato durante la evaporación. El residuo de esta evaporación se trata con ácido fenoldisulfónico y se alcaliniza con NH4OH. Entonces se mide la intensidad del color amarillo que se produce como resultado de estos tratamientos.
El método es aplicable al extracto de saturación del suelo. En cualquier caso, es necesario evaporar a sequedad porque la reacción del ácido fenoldisulfónico con los nitratos es afectada por el agua.
20.2. Material y aparatos
Colorímetro con un recorrido de luz de 1 cm. y equipado con filtro azul que tenga un máximo de transmitancia de 420 mμ o espectrofotómetro.
20.3. Reactivos
20.3.1. Sulfato cálcico en polvo (S04Ca. 2H2O).
20.3.2. Acido fenoldisulfónico (fenol 2: 4-ácido disulfónico.)
Disolver 25 g. de fenol puro en 150 ml. de SO4H2 concentrado (p. e. 1,84) en un matraz erlenmeyer de 500 ml. A continuación añadir 75 ml. de SO4H2 humeante (13 a 15 SO3), mezclar la solución y colocar el matraz en agua hirviendo durante dos horas. Almacenar la solución resultante de ácido fenoldisulfónico en un frasco topacio con tapón de cristal. O bien disolver 25 g. de fenol puro en 225 ml. de SO4H2 concentrado (p. e. 1,84), colocando el matraz erlenmeyer en agua hirviendo durante seis horas.
20.3.3. Solución de hidróxido amónico (NH4OH). Preparar este reactivo mezclando 1 litro de NH4OH (p. e. 0,89 a 0,90) con 1 litro de agua.
20.3.4. Carbonato cálcico en polvo.
20.3.5. Solución patrón de nitrato: disolver 0,722 g. de NO3K en agua, y diluir la solución en un volumen de 1.000 ml. en un matraz aforado. Si se usa NO3K puro y seco, esta solución contiene 100 µg. de N en forma de nitrato por ml. Almacenar la solución en la nevera.
20.3.6. Sulfato de plata (SO4Ag2) en polvo.
20.3.7. Hidróxido cálcico [Ca(OH)2] en polvo.
20.3.8. Carbonato magnésico (CO3Mg) en polvo.
20.4. Procedimiento
20.4.1. Preparación del extracto de suelo. Colocar 50 g. de suelo en un frasco de cuello ancho de 500 ml. y añadir 0,5 g. de CO3Ca. 2H2O y 250 ml. de agua. Tapar y agitar durante diez minutos en un agitador mecánico. Dejar sedimentar durante algunos minutos, y decantar y filtrar el líquido sobrenadante a través de un papel de filtro grande plegado (Whatman número 42). Si la primera porción de filtrado no es clara, volver a filtrar y recoger el filtrado en un frasco limpio.
20.4.2. Para suelos que contienen menos de 15 p. p. m. de cloruros. Pipetar una alícuota de 25 ml. del extracto en un vaso pyrex de 150 ml., añadir 0,05 g. de CO3Ca y evaporar la muestra a sequedad en una placa de calentamiento. Llevar a cabo la evaporación en una atmósfera libre de vapores de NO3H y no continuar el calentamiento una vez que se haya alcanzado la sequedad. Dejar enfriar el vaso, añadir 2 ml de ácido fenoldisulfónico con una pipeta que vierta rápidamente. Rotar el vaso de manera que el ácido fenoldisulfónico se ponga en contacto con todo el residuo. Dejar reposar durante diez minutos, añadir 20 ml de agua y agitar con una varilla de vidrio hasta que todo el residuo se disuelva. Entonces añadir con una bureta despacio y removiendo solución NH4OH hasta que la solución sea ligeramente alcalina (cuando aparezca el color amarillo), y añadir 2 ml. más para asegurar el exceso de este reactivo. Transferir la solución a un matraz aforado de 100 ml. y diluir con agua. Mezclar bien y medir la intensidad del color con luz de 420 mµ comparando con la absorción que se produce por un ensayo en blanco.
Determinar el contenido de nitratos de la muestra usando un gráfico de calibración que se construye con los resultados obtenidos con muestras que contengan 0,10, 20, 40, 70 y 100 µg de N en forma de nitrato. Para preparar este gráfico, diluir 10 ml. de la solución patrón de nitratos (reactivo 20.3.5) con agua en un matraz aforado de 100 ml. y mezclar bien. Añadir alícuotas de 0, 1, 2, 4, 7 y 10 ml. de esta solución patrón diluidas mediante una bureta de 10 ml. dividida en intervalos de 0,02 ml. a vasos pyrex de 150 ml. y medir la intensidad de los colores que se obtienen con estas muestras según se describe en el análisis del extracto del suelo.
20.4.3. Para suelos que contienen más de 15 p. p. m. de cloruros. El procedimiento es el mismo que se describe en 20.4.2, excepto que se utiliza una alícuota del extracto en la que se hayan eliminado los cloruros. Para ello, añadir 0,1 g. de SO4Ag2 a 100 ml. del extracto en un erlenmeyer de 250 ml. y agitar durante quince minutos. Añadir entonces 0,2 g. de Ca(OH)2 y 0,5 g. de CO3 Mg para precipitar el Ag+, agitar el matraz durante cinco minutos. Filtrar la suspensión a través de un papel de filtro seco y plegado, y analizar el filtrado por el procedimiento descrito en 20.4.2 (puede omitirse la adición de CO3Ca antes de la evaporación).
20.5. Cálculo
Calcular el N de nitrato expresado en meq/l. del extracto.

P = peso en µg de N nitratos deducido de la curva de calibración.
V = volumen en ml. de la alícuota del extracto.
20.6. Observaciones
Los extractos deben de analizarse inmediatamente después de su preparación, almacenándolos en una nevera cuando ello no sea posible.
Se recomienda llevar a cabo la evaporación de sequedad en baño de agua.
La solución patrón de nitratos es estable durante seis meses como mínimo cuando se conserva en una nevera.
20.7. Referencia
1. Bremmer, J. M.: «Methods of Soil Analysis». Part 2: 1.215-1.219. American Society of Agronomy, 1965.
21. BORO. COLORIMETRIA CON CURCUMINA
(Provisional)
21.1. Principio
El método propuesto se basa en la formación de rosacianina, que tiene lugar en forma proporcional a la cantidad de boro presente. La presencia de ácido oxálico mantiene la acidez a un nivel suficiente para que se desarrolle el color si la disolución que contiene el boro es ligeramente acida, con lo cual queda eliminada la necesidad de añadir Cl H concentrado.
21.2. Material y aparatos
21.2.1. Vaso de vidrio exento de boro de 250 ml.
21.2.2. Matraces aforados de 1.000 ml., 500 ml. y 50 ml.
21.2.3. Tubos colorimétricos normalizados.
21.2.4. Fotocolorímetro.
21.3. Reactivos
21.3.1. Etanol al 95 por 100, de calidad reactivo, que deberá redestilarse si es necesario para que quede exento de boro.
21.3.2. Solución de curcumina-ácido oxálico. Disolver en 100 ml. de etanol del 95 por 100 exento de boro, 0,04 g. de curcumina finamente pulverizada y 5 g. de ácido exálico (C2O4H2. 2H2O). Esta solución debe de almacenarse en una botella topacio o de plástico de color ámbar en un lugar oscuro y fresco. Debido a la descomposición de la curcumina, este reactivo debe de prepararse de nuevo cada varios días. El plazo de conservación puede extenderse hasta una semana o más si se mantiene en un frigorífico.
21.3.3. Solución patrón de boro. Disolver 0,572 g. de BO3H3 desecado, de calidad reactivo, en 1 litro de agua, con lo que se obtiene un patrón de 100 μg de B por ml. Preparar una solución diluida tomando, mediante una pipeta, 50 ml. de la primera disolución patrón, pasándola a un matraz aforado de 500 ml. y enrasando con agua destilada, con lo que se obtiene un patrón de 10 μg de B por ml. de volumen. La disolución patrón diluida se emplea para preparar una serle de patrones en matraces aforados de 50 ml. para construir las curvas de calibración de acuerdo con la tabla núm. 21.1.
21.4. Procedimiento
Transferir una parte alícuota de 1 ml. de una disolución ligeramente ácida, que contenga entre 0,2 y 5 μg de boro a un vaso de vidrio exento de boro de 250 ml. de capacidad. La misma pipeta de 1 ml. se emplea para transferir el patrón y las disoluciones a ensayar y, por tanto, no es necesario establecer un calibrado volumétrico especial; sin embargo, el volumen debe de mantenerse igual a 1 ml. A continuación de esto, añadir al vaso 4 ml. de solución de curcumina-ácido oxálico y mezclar dando vueltas al vaso. Finalmente, evaporar la solución a sequedad en una estufa (o en un baño de agua) regulada a 55° ± 3° C, manteniendo el residuo a esta temperatura durante quince minutos, para asegurar que quede completamente seco. La sustancia coloreada, rosacianina, se desarrolla durante la evaporación y desecación.
Enfriar el vaso que contiene el residuo seco hasta la temperatura ambiente. Seguidamente añadir 25 ml. de etanol y triturar el residuo para extraer el compuesto colorante. Filtrar la disolución a través de un papel de filtro Whatman núm. 2, recogiendo el filtrado directamente en el tubo del colorímetro (la ligera contaminación de boro procedente del papel de filtro no afecta la medida, pues el pase de formación de color ya ha sido superado).
Hacer la lectura a 540 mμ dentro de las dos primeras horas, puesto que la rosacianina se hidroliza gradualmente con formación de curcumina; esto se aprecia al cabo de dos horas. Si el tanto por ciento de transmisión fuera demasiado bajo (menos de un 25 por 100 a 30 por 100), lo que indicaría la presencia de más de 1,5μg de B, volver a leer inmediatamente la disolución a 580 ó 600 mμ.
21.5. Cálculo
Hacer la lectura en la curva de calibración correspondiente a la longitud de onda utilizada para la determinación y, de esta forma, se determinan los μg de boro contenidos en la alícuota de 1 ml.
21.6. Observaciones
Los nitratos en cantidad superior a 20 μg de N en 1 ml. de alícuota interfieren en el análisis de la disolución ácida. En este caso se pueden eliminar de la disolución mediante evaporación de una parte alícuota del problema a la que se le haya añadido una cantidad suficiente de agua saturada con Ca (OH)2 para volverla alcalina, sometiendo el residuo a una ignición suave. A continuación disolver la ceniza en CIH 0,05 N.
También interfieren en la determinación de boro los iones Fe, Mo, Ti y Zr si se encuentran en proporciones más altas de lo normal (por encima de unas 300 p. p. m.) en la disolución problema.
Cuando la parte alícuota de 1 ml. contenga una cantidad insuficiente de boro, es posible concentrar la disolución por evaporación, después de haberla alcalinizado con disolución de Ca (OH)2, disolviéndola, seguidamente, en un pequeño volumen de CIH de concentración suficiente para obtener una disolución ligeramente ácida.
21.7. Referencias
1. Dible, W. T.; Troug, E., y Berger, K. C.: «Boron determination in soils and plants. Simplified curcumin procedure». Anal. Chem. 26; 418-421, 1954.
2. Jackson, M. L.: «Análisis químico de suelos». 503-506. Edit. Omega, 1964.
TABLA 21.I.
Escala de concentraciones de las soluciones patrón de boro para el método de curcumina
22(a) YESO
(Cualitativo)
22(a).1. Principio
Formación de precipitado al añadir acetona a un extracto acuoso de suelo.
22(a).2. Material y aparatos
22(a).2.1. Agitador mecánico.
22(a).3. Reactivos
22(a).3.1. Acetona.
22(a).4. Procedimiento
Pesar una muestra de suelo desecado al aire de 10 a 20 g. y colocarla en un frasco al que se añade agua suficiente para disolver parta o todo el yeso presente. Tapar la botella y agitar a mano seis veces a intervalos de quince minutos o durante quince minutos en agitador mecánico. Filtrar el extracto a través de un filtro de porosidad media. Colocar unos 5 ml. del extracto en un tubo de ensayo y añadir aproximadamente un volumen igual de acetona. La formación de un precipitado indica la presencia de yeso en el suelo.
22(a).5. Observaciones
El suelo no debe ser desecado en estufa debido a que el calentamiento del SO4 Ca 2H2O promueve su conversión en SO4 Ca 0,5H2O. Este hidrato tiene una solubilidad en agua 20 por 100 superior al yeso durante un periodo indefinido después de su disolución.
22(a).6. Referencia
1. United States Salinity Laboratory Staff: «Diagnosis and Improvement of Satine and Alkali Soils»: p. 103, 1954.
22(b) YESO
(Cuantitativo)
22(b).1. Principio
En caso de reacción positiva del método 22(a), puede interesar conocer la cantidad de yeso presente. Se basa este método en que la solución del sulfato cálcico en acetona es menor que la de otros sulfatos generalmente menos abundantes en el suelo. El precipitado que se obtiene por adición de acetona al extracto acuoso del suelo es redisuelto en agua, midiéndose la conductividad eléctrica de dicha disolución para determinar la concentración de sulfato cálcico presente.
22(b).2. Material y aparatos
22(b).2.1. Centrífuga.
22(b).2.2. Tubos de centrífuga de fondo cónico de 50 ml.
22(b).2.3. Celda de conductividad.
22(b).2.4. Puente de Wheatstone, con un indicador catódico del tipo ojo mágico (ver 7.2.1).
22(b).3. Reactivos
22(b).3.1. Acetona.
22(b).4. Procedimiento
Pesar de 10 a 20 g. de suelo desecado al aire y colocar en un frasco de unos 250 ml. Añadir un volumen de agua de 5 a 10 veces superior al peso de suelo. Tapar el frasco y agitar a mano seis veces, cada quince minutos, o en un agitador mecánico durante quince minutos. Filtrar el extracto a través de papel de filtro de porosidad media. Transferir 20 ml. del extracto obtenido a un tubo [22(b).2.2] de centrífuga y agregar 20 ml. de acetona y mezclar. Dejar en reposo durante cinco o diez minutos hasta que flocule el precipitado. Centrifugar con una fuerza centrífuga relativa de 1.000 durante tres minutos. Decantar el líquido sobrenadante, invertir el tubo y drenar sobre papel de filtro durante cinco minutos. Dispersar el precipitado y enjuagar la pared del tubo con un chorro de acetona de 10 ml. soplando por una pipeta. Repetir la centrifugación, decantación y escurrido como antes. Finalmente añadir 40 ml. de agua destilada al tubo, tapar y agitar hasta que el precipitado quede completamente disuelto. Medir la conductividad eléctrica de la solución según se describe en el método 7 y expresar la conductividad a 25° C usando el factor de corrección apropiado (tabla 7.1).
22(b).5. Cálculo
Calcular el contenido en yeso del suelo expresado en porcentaje.

C = concentración en meq/l. de SO4Ca 2H2O deducida de la lectura de conductividad en la figura 22(b).I.
V' = volumen en ml. de agua añadida para disolver el precipitado.
P = peso en g. de la muestra de suelo.
V = volumen en ml. del agua añadida a la muestra de suelo.
V'' = volumen en ml. del extracto acuoso utilizado para precipitar con acetona.
22(b).6. Observaciones
22(b).6.1. El suelo no debe de desecarse en estufa [22(a).5].
22(b).6.2. Los sulfatos sódicos y potásicos cuando están presentes en concentraciones altas también son precipitado, por la acetona. Las concentraciones máximas de sulfatos sódicos y potásicos permisibles son de 50 y 10 meq/l., respectivamente.
22(b).6.3. Si habiendo empleado una relación de suelo : agua de 1 : 5 se encuentra después de los cálculos que el suelo contiene unos 15 meq. de yeso por 100 g. de suelo (o 1,29 por 100) hay que repetir la extracción y determinación usando una relación suelo : agua más baja (1 : 10 a 1 : 100). Con la relación 1 : 100 se podrá determinar el yeso total incluso en suelos que contengan un 25 por 100 de yeso.
22(b).7. Referencia
1. United States Salinity Laboratory Staff: «Diagnosis and Improvement of Saline and Alkali Soils»: p. 104, 1954.
TABLA 22(b).I.
Datos para construir la figura 22(b).I
| Concentración de S04Ca en: meq/l. | Conductividad eléctrica de la solución en: mmhos/cm. |
|---|---|
| 1 | 0,121 |
| 2 | 0,226 |
| 5 | 0,500 |
| 10 | 0,900 |
| 20 | 1,584 |
| 30,5 | 2,205 |
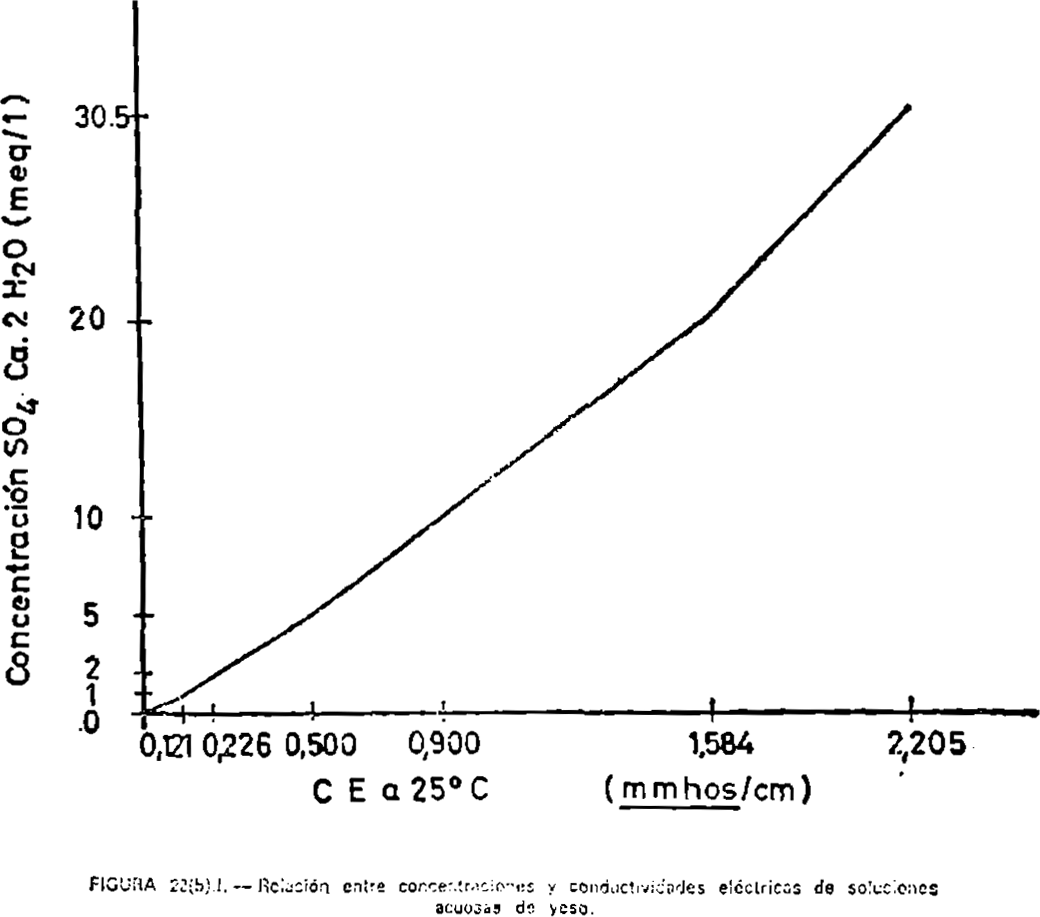
23(a) NECESIDAD DE YESO
23(a).1. Principio
Puesto que un suelo alcalino es el que tiene un exceso de Na absorbido, si una solución saturada de yeso se pone en contacto en un frasco con una muestra del suelo después de un cierto tiempo se alcanzará el equilibrio. La determinación en la solución de equilibrio del Ca + Mg (método 16) y sustracción de este valor a la cantidad de Ca presente en la solución acuosa de yeso se tendrá una estimación del Ca que ha desplazado parte del Na absorbido y consecuentemente un valor aproximado de la necesidad del yeso del suelo. El método es el de Schoonover (1952) tal como es descrito por el United States Salinity Laboratory Staff (1954).
23(a).2. Material y aparatos
23(a).2.1. Microbureta de 10 ml.
23(a).2.2. Erlenmeyer de 125 ml.
23(a).2.3. Agitador mecánico de frascos.
23(a).2.4. Frasco de 150 ó 200 ml.
23(a).3. Reactivos
23(a).3. (1, 2, 3, 4, 5 y 6) como 16.3. (1, 2, 3, 4, 5 y 6).
23(a).3.7. Solución acuosa de yeso aproximadamente saturada, de concentración de Ca conocida. Colocar unos 5 g. de yeso (SO4Ca 2 H2O) en un frasco con 1 litro de agua, tapar y agitar a mano varías veces en el intervalo de una hora o en un agitador mecánico durante diez minutos. Filtrar y determinar la concentración, de calcio en una alícuota de 5 ml. tal como se describe en 16.4.2. La concentración total de calcio debe ser al menos 28 meq/l.
23(a).4. Procedimiento
Pesar 5 g. de suelo desecado al aire y colocar en un frasco al que se le añaden 100 ml. de reactivo [23(a).3.7] medidos con una pipeta. Tapar el frasco y agitar a mano cada cinco minutos durante un tiempo de treinta minutos o durante cinco minutos en un agitador mecánico. Filtrar la suspensión y determinar la concentración de calcio + magnesio en una alícuota apropiada tal como se describe en 16.4.3.
23(a).5. Cálculo
Calcular la necesidad de yeso expresada en meq/100 g. y en Tm/Ha. x 15 cm.
Necesidad de yeso, meq/100 g. = 2 (C ‒ C').
Necesidad de yeso, Tm/Ha. x 15 cm. = 1,677·2· (C ‒ C').
C = concentración en meq/l. de Ca en la solución original de yeso.
C' = concentración en meq/l. de Ca + Mg en el filtrado.
23(a).6. Observaciones
Debido a la naturaleza empírica del método en muchos suelos no se ha encontrado buena correlación entre el porcentaje de sodio cambiable (PSC), la necesidad de yeso tal como se calcula por este método y el contenido en yeso del suelo.
23(a).7. Referencia
1. United States Salinity Laboratory Staff: «Diagnosis and Improvement of Saline and Alkali Soils»: p. 104, 1954.
23(b) CALCULO DE LA NECESIDAD DE ENMIENDA
23(b).1. Principio
Se requiere conocer: 1.°) la capacidad de cambio catiónico (CCC).en meq/100 g. de suelo determinada según se describe en el método 9; 2.°) la cantidad de sodio de cambio (SC) en meq/100 g. de suelo, bien por determinación directa por el método 10(a) o por su estimación indirecta de los datos analíticos de cationes solubles (Na, Ca y Mg) en el extracto de saturación del suelo [método 13(a), 14, 15 y 16], siguiendo las instrucciones que se dan en el siguiente procedimiento de cálculo.
23(b).2. Cálculo
23(b).2.1. En caso de que no se haya hecho la determinación del sodio, pero si el análisis del extracto de saturación del suelo, estimar aquel (SC) de las concentraciones de Na, Ca y Mg en el extracto, comenzando por calcular la relación de absorción de sodio (RAS).

extracto de saturación en meq/l. Para ahorrar éste cálculo utilizar el nomograma de la figura 23(b).1, uniendo mediante una regla la concentración de Na en el eje de la izquierda con la de Ca + Mg en el eje de la derecha, leyendo el valor del RAS en la intersección de dicha recta con la recta inclinada del nomograma. Al mismo tiempo se lee el PSCi (porcentaje de sodio cambiable inicial) en la parte inferior de la recta inclinada del nomograma.
23(b).2.2. En caso de haberse determinado directamente el S Ci (sodio de cambio inicial del suelo) calcular el P S Ci, por la fórmula

23(b).2.3. Decidir el porcentaje de sodio de cambio que se va a tolerar cuando el suelo se haya recuperado (P S C f) que normalmente debe de oscilar entre 0 por 100 y 10 por 100. A continuación calcular la cantidad de sodio que hay que reemplazar (S R) para alcanzar ese porcentaje de sodio de cambio final, de acuerdo con la fórmula

23(b).2.4. En la tabla 23(b).I, se dan las cantidades de enmienda necesarias por Ha. para reemplazar la cantidad de sodio indicada, en una profundidad de suelo de 15 a 30 cm y bajo el supuesto de que el desplazamiento fuese cuantitativo.
23(b).2.5. En la tabla 23 (b).II se dan las cantidades de otras enmiendas que pueden añadirse a los suelos alcalinos equivalentes a Tm. de azufre.
23(b). 3. Observaciones
La reacción entré una enmienda tal como el yeso y el sodio de cambio es naturalmente una reacción de equilibrio y, por tanto, no se produce reemplazamiento cuantitativo del sodio por el calcio añadido. Como regla general se sugiere para compensar esta falta de reemplazamiento cuantitativo multiplicar las cantidades de enmiendas indicadas en la tabla 23(b).I por un factor de 1,25.
23(b).4. Referencia
1. United States Salinity Laboratory Staff: «Diagnosis and Improvement of Saline and Alkali Soils»: pp. 49-50, 1954.
TABLA 23(b).I.
Cantidades de yeso y azufre requeridas para reemplazar las cantidades indicadas de sodio de cambio (S C)
| Sodio de cambio a reemplazar meq/100 g. de suelo | Yeso SO4Ca. 2H2O Tm/Ha. x 30 cm. | Yeso SO4Ca. 2H2O Tm/Ha. X 15 cm. | Azufre (1) S Tm/Ha. X 30 cm. | Azufre (1) S Tm/Ha. X 15 cm. |
|---|---|---|---|---|
| 1 | 3,8 | 1.9 | 0,72 | 0,36 |
| 2 | 7,6 | 3.8 | 1,43 | 0,72 |
| 3 | 11,6 | 5,8 | 2,15 | 1,08 |
| 4 | 15,5 | 7,8 | 2,87 | 1,44 |
| 5 | 19,3 | 9,7 | 3,59 | 1,80 |
| 6 | 23,1 | 11,6 | 4,30 | 2,15 |
| 7 | 26,9 | 13,5 | 5,02 | 2,51 |
| 8 | 30,7 | 15,4 | 5,74 | 2,87 |
| 9 | 34,7 | 17.4 | 6,46 | 3,23 |
| 10 | 38,6 | 19,3 | 7,17 | 3,59 |
|
(1) Enmienda recomendable tan sólo en suelos alcalinos calizos. |
||||
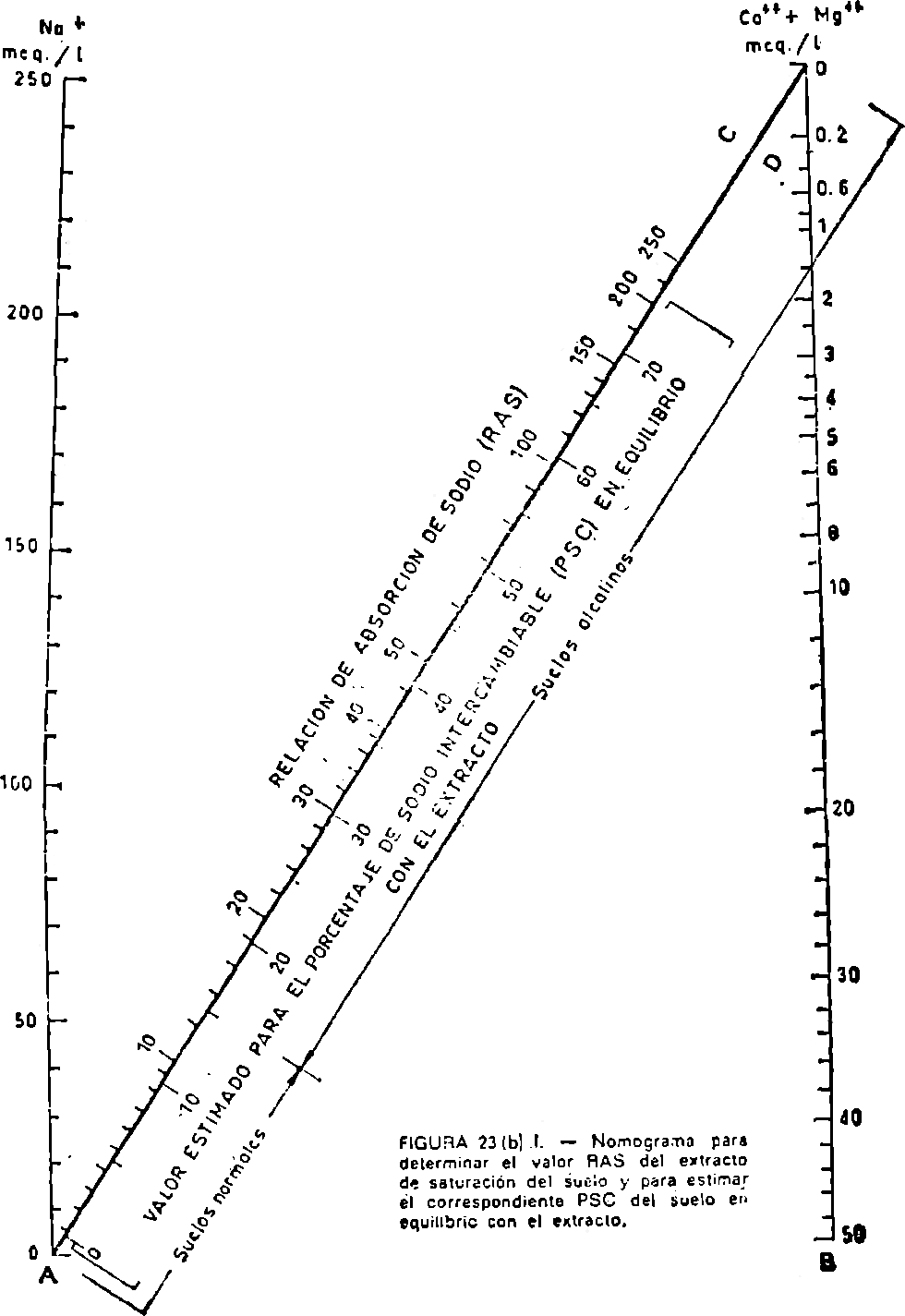
TABLA 23(b).II.
Equivalencia en azufre de las distintas enmiendas
| Enmienda | Tm. equivalente a 1 Tm. de azufre |
|---|---|
| Azufre. | 1,00 |
| Solución de cal-azufre (24 por 100 de S). | 4,17 |
| Acido sulfúrico. | 3,06 |
| Yeso (SO4 Ca. 2 H2O). | 5,38 |
| Sulfato de hierro (SO4 Fe. 7 H2O). | 8,69 |
| Sulfato de aluminio [(SO4)3Al2. 18 H2O]. | 6,94 |
| Caliza (CO3 Ca). | 3,13 |
24. CALIZA ACTIVA
(Provisional)
24.1. Principio
La caliza activa se calcula por una dosificación gasométrica de CO2 del carbonato amónico, formado al reaccionar el carbonato cálcico activo con el oxalato amónico en disolución.
Con este método es posible la apreciación del carbonato de calcio activo aun en suelos que contienen yeso y materia orgánica.
24.2. Material y aparatos
Calcímetro Bernard. Como en 3.2.1.
24.3. Reactivos
24.3.1. Solución de oxalato amónico, aproximadamente 0,2 N. Disolver 14,212 g. de oxalato amónico en 1 litro de agua.
24.3.2. Acido clorhídrico 1 : 1 (aproximadamente 6 N).
24.3.3. Carbonato cálcico puro pulverizado.
24.4. Procedimiento
Tomar una muestra de suelo desecado al aire, pasarla por un tamiz de 2 mm. de abertura de malla y pesar, según sea el contenido en caliza de la muestra, de 2,5 a 10 g. con una precisión de 1 mg. Introducir el suelo en un erlenmeyer de 500 ml. con tapón esmerilado.
Añadir exactamente 250 ml. de la solución de oxalato amónico y agitar mecánicamente durante dos horas. Filtrar desechando los primeros filtrados.
Tomar 25 ml. del filtrado y proceder con el ácido clorhídrico por el método del calcímetro de Bernard, como en 3.4, anotando el volumen de CO2 desprendido.
Para tener en cuenta la influencia de la presión y la temperatura en el momento de la determinación, proceder como anteriormente utilizando 0,10 g. de carbonato cálcico puro pulverizado, anotando el volumen de anhídrido carbónico desprendido.
24.5. Cálculo
Expresar el contenido en caliza activa en tanto por mil

C = tanto por mil de caliza activa.
P = peso en g. de la muestra.
V = volumen de anhídrido carbónico desprendido por el extracto de la muestra.
V' = volumen de anhídrido carbónico desprendido por el carbonato cálcico puro.
24.6. Referencias
1. Drouineau, G: «Guide pour l’étude experimentale du sol». París, 1952.
2. Nijensohn, L., y Pizarro, O. C.: «Un procedimiento para la determinación del calcáreo activo en suelos orgánico-yesosos». Boletín Técnico núm. 2. Instituto Provincial Agropecuario, 1960.
25. CARBONO ORGANICO OXIDABLE
25.1. Principio
En el presente método se determina el carbono orgánico del suelo que se oxida con dicromato en presencia de ácido sulfúrico E exceso de oxidante se valora con sulfato ferroso amónico (sal de Mohr) y la cantidad de carbono orgánico oxidado se calcula a partir de la cantidad de dicromato reducido.
La presencia en el suelo de otras sustancias, oxidables o reducibles, pueden dar contenidos en carbono orgánico altos o bajos, respectivamente.
25.2. Material y aparatos
25.2.1. Matraces erlenmeyer de 500 ml.
25.2.2. Barras agitadoras.
25.2.3. Agitador magnético y buretas o potenciógrafo automático con sus correspondientes buretas.
25.3. Reactivos
25.3.1. Dicromato potásico 1 N. Disolver 49,05 g. de dicromato potásico en agua y diluir hasta un litro.
25.3.2. Acido sulfúrico concentrado conteniendo 25 g. de SO4Ag2 por litro.
25.3.3. Acido fosfórico concentrado.
25.3.4. Difenilamina en solución sulfúrica. Disolver 0,5 g. de difenilamina en 20 ml. de agua y añadir 100 ml. de ácido sulfúrico concentrado. Si se dispone de potenciógrafo automático no se precisa el indicador citado anteriormente.
25.3.5. Sulfato ferroso amónico (Sal de Mohr), 0,5 N. Disolver 196,1 g. de (SO4)2 Fe (NH4)2 6H2O en 800 ml. de agua que contenga 20 ml. de ácido sulfúrico concentrado, diluyendo hasta 1 litro. Normalizar esta solución cada vez que se emplee, valorándola, con 10 ml. de solución 1 N de dicromato potásico (25.3.1), siguiendo en todo el procedimiento que se indica en 25.4.
25.4. Procedimiento
Pesar una muestra de tierra fina triturada del orden de 0,5 g. (0,05 g. en el caso de turba; 2 g. para suelos que contengan menos del 0,5 por 100 de carbono orgánico oxidable) y colocarla en un matraz erlenmeyer de 500 ml. Añadir, mediante bureta, 10 ml. de reactivo 25.3.1 imprimiendo un movimiento de giro al matraz para asegurar una mezcla íntima con el suelo. Añadir, lentamente y agitando, 20 ml. de reactivo 25.3.2, dejando la mezcla en reposo durante treinta minutos sobre una placa de amianto. Añadir 200 ml. de agua, dejar enfriar y agregar, a continuación, 10 ml. del reactivo 25.3.3., introduciendo también en el matraz una barra agitadora con recubrimiento de teflón.
Si no se dispone de potenciógrafo automático, añadir. 4 ó 5 gotas del indicador 25.3.4 y valorar con 25.3.5 mediante bureta y utilizando agitador magnético hasta cambio de color. La coloración vira de rojo burdeos a verde brillante, pasando por tonos azul violáceos.
Si se dispone de potenciógrafo, valorar automáticamente con 25.3.5 en escala de milivoltios y utilizando electrodo de placa de platino frente a electrodo de referencia de sulfato de mercurio.
Cuando se reduce más del 80 por 100 del dicromato, la determinación debe repetirse con una muestra de suelo más pequeña.
La adición de sulfato de plata al ácido (25.3.2), se hace para eliminar la posible interferencia de cloruros, la adición de ácido fosfórico, tiene por objeto favorecer el progreso de la reacción de valoración por bloqueo del ion ferroso, se minimiza por operar con muestras aireadas. La interferencia del Mn no parece causar, en las circunstancias más desfavorables, un error superior al 6 por 100.
25.5. Cálculo
Calcular el carbono orgánico oxidable, expresado en porcentaje de tierra fina seca a 105° C. mediante la fórmula

M = miliequivalentes de dicromato potásico añadidos.
M' = miliequivalentes de sulfato ferroso amónico consumidos.
P = peso, en gramos, de la muestra referido a peso seco a 105° C.
25.6. Observaciones
Para expresar el resultado en porcentaje de materia orgánica oxidable se multiplica por 1,72 (coeficiente de Waksman).
25.7. Referencias
1. Hesse, P. R.: «A Textbook of soil Chemical analysis». John Murray. London, 1971.
2. Jackson, M. L.: «Análisis químico de suelos» Omega. Barcelona, 1964.
3. Walkey, A.: «An examination of methods for determining organic carbon and nitrogen in soils». Jour Agr. Sci. 25: pp. 598-609, 1935.
Métodos físicos
1. HUMEDAD
1.1. Procedimiento
Colocar una muestra representativa del suelo en recipiente adecuado previamente tarado. Es deseable, siempre que sea posible, tomar al menos una muestra de 25 g. Pesar y secar sin tapar hasta peso constante en una estufa a 105° C. Poner a enfriar el recipiente tapado en un desecador y pesar de nuevo.
1.2. Material y aparatos
1.2.1. Estufa de desecación con regulación de temperatura hasta 150° C.
1.2.2. Desecadores.
1.2.3. Recipientes para contener muestras de suelo que pueden ser latas cilíndricas con tapa que cierre a presión.
1.3. Cálculo
Calcular el contenido en humedad expresado en porcentaje

P = peso en g. de la muestra de suelo.
P' = peso en g. de la muestra de suelo seco.
1.4. Observaciones
El tiempo necesario para alcanzar peso constante (el término no debe de interpretarse en sentido estricto, ya que un peso constante rara vez se alcanza, excepto en suelos muy arenosos que contengan poca materia orgánica) dependerá del tipo de estufa que se use, del tamaño y profundidad de la muestra y del tipo de suelo. Si se usa estufa con corriente de aire forzada, normalmente bastará con diez horas. Si la estufa es de convección, las muestras deben desecarse durante veinticuatro horas como mínimo y se deben de tomar precauciones para no introducir muestras húmedas durante la última mitad del período de secado.
Cuando no se requiera gran precisión, puede utilizarse lámpara infrarroja generalmente incorporada a una balanza para desecar la muestra, pudiéndose en este caso colocar el suelo en una cápsula de tara conocida.
2(a) TEXTURA
(Densímetro Bouyoucos)
2(a).1. Principio
Los métodos ordinarios de determinación de la textura de los suelos requieren que las partículas estén dispersadas en una solución acuosa. La agitación del suelo en una solución alcalina diluida de metafosfato sódico es suficiente, en muchos casos, para la dispersión de todos los agregados del suelo. Sin embargo, los suelos que contienen cantidades considerables de sales fácilmente solubles, tal como yeso, o materia orgánica, puede resistir la dispersión, al menos que se eliminen previamente dichos componentes. La eliminación de la materia orgánica puede realizarse tratando el suelo con peróxido de hidrógeno y subsecuente filtración y lavado con suficiente agua para disolver el yeso.
La efectividad de los agentes dispersantes depende de la absorción de sodio que desplace a otros cationes absorbidos con el consiguiente desarrollo de fuerzas eléctricas de repulsión entre las partículas del suelo. Las mezclas de metafosfato y carbonato sódico son particularmente efectivas y han hecho posible la dispersión de suelos calizos sin eliminación previa de los carbonatos alcalino-térreos.
Mediante el densímetro se mide la densidad de la suspensión del suelo (φ) que está relacionada con la concentración de partículas en dicha suspensión (c) mediante la fórmula

en donde P1 y Ps son las densidades del líquido y de las partículas, respectivamente La escala del densímetro puede estar calibrada en unidades de concentración, para valores dados de P1 y Ps.
El método del densímetro, como el método de la pipeta (2 b), está basado en la ecuación de Stokes:

que relaciona el diámetro de las partículas (x) con el tiempo de caída (t) y un parámetro Ө que no es constante durante el proceso de sedimentación, ya que la profundidad de inmersión del densímetro es variable, «h» representa el espesor vertical de la zona a partir de la superficie de la suspensión que está libre de partículas inferiores a un determinado diámetro (x) después de un tiempo de sedimentación t. Igualando h a la distancia en centímetros desde la superficie de la suspensión hasta el centro del bulbo del densímetro, es posible relacionar los valores de h con las lecturas del densímetro L, haciendo, Ө una función de L. Esta relación se da en la tabla 2(a).I para el densímetro que se recomienda en 2(a).2. Dicha tabla está basada en la ecuación de Stokes, usando los siguientes valores de las constantes:
ƞ = ƞ30 = viscosidad del agua a 30° C = 0,008007 poise.
P1 = densidad de una solución de Calgon al 0,5 por 100 = 0,99949 g/ml.
Ps = densidad de la partícula = 2,650 g/ml.
g = 980,7 cm/seg2.
La constante 1.000 es el factor de conversión del diámetro de las partículas en mm. a micras.
2(a).2. Material y aparatos
2(a).2.1. Densímetro ASTM núm. 152 H, con la escala de Bouyoucos en g/l. (American Society Testing Material).
2(a).2.2. Batidora eléctrica con paletas recambiables.
2(a).2.3. Probeta graduada de 1 litro con la señal de enrase de 1.000 ml. a unos 36 cm. del fondo.
2(a).2.4. Embolo agitador del latón. En el centro de un disco de 2 mm. de espesor y de 50 mm. de diámetro, soldar una varilla metálica de 50 cm. de longitud y 5 mm. de diámetro.
2(a).3. Reactivo
2(a).3.1. Disolver 50 g. de «calgon» en agua y completar hasta 1 litro. Calgon es una preparación comercial de metafosfato sódico (PO3Na) que contiene la cantidad apropiada de CO3Na2 como para dar un pH de 8,3 en solución acuosa al 10 por 100. La solución recomendada tiene una concentración ligeramente superior a 0,5 N de sodio.
2(a).4. Procedimiento
Calibrar cada densímetro de la siguiente forma: añadir 100 ml. de la solución de calgon a la probeta y agregar agua destilada hasta enrasar la señal de 1 litro. Mezclar la solución con el émbolo agitador y esperar hasta que alcance la temperatura ambiente. Tomar la temperatura con cuidado, sumergir el densímetro en la solución y determinar la lectura de la escala en el borde superior del menisco que rodea el vástago del densímetro. Cuando se trabaja con suspensiones de suelos no es posible hacer la lectura mirando el vástago por debajo de la superficie del líquido. Tanto en la calibración del hidrómetro como en las medidas de suspensión debe de adoptarse el siguiente método para efectuar la lectura: La posición del menisco se puede determinar con precisión mirándolo desde un ángulo de 10 a 20° por encima del plano del liquido, fijándose en la línea horizontal brillante (imagen de difracción) formada en la escala por una lamparita que se mantiene opuesta a la frente y protegiendo los ojos de la luz directa con una pantalla. A veces, debido a la falta de limpieza del vástago en presencia de sustancias grasas del suelo, la línea brillante puede no aparecer, porque el menisco no forma el ángulo de contacto normal de cero grados. Cuando esto ocurra, se levantará y bajará el densímetro ligeramente en la suspensión hasta que se forme el ángulo de contacto normal.
Pesar 40,0 g. de suelo para la determinación de la textura y una cantidad similar para la determinación del peso seco, en caso de que no se conociese la humedad de la muestra secada al aire según se determina por el método 1. Desecar la segunda muestra en una estufa a 105° durante la noche y pesarla de nuevo. Colocar la primera muestra en un vaso de 600 ml. Añadir 100 ml. de la solución de calgon y unos 400 ml. de agua destilada y dejar que se empape durante un mínimo de diez minutos.
Transferir la suspensión al vaso dispersador de la batidora usando el chorro de un frasco lavador con agua destilada para arrastrar toda la muestra. Batir la suspensión durante cinco minutos y transferir a la probeta, enrasando con agua destilada hasta la señal de 1.000 ml.
Colocar la probeta en el banco de sedimentación. Anotar la temperatura de la suspensión cuando se estabilice. Introducir el émbolo en la probeta y moverlo de arriba abajo para mezclar bien la suspensión. Sostener la probeta firmemente con la mano libre cuando se tire del émbolo hacia arriba. El movimiento de éste en las proximidades de la superficie libre debe de ser suave para no derramar el contenido. Dar emboladas fuertes cuando se tire del émbolo en las proximidades del fondo para arrastrar cualquier partícula que haya podido depositarse en él. Desalojar los sedimentos depositados en los bordes inferiores del fondo del cilindro inclinando la varilla del agitador e imprimiéndole un movimiento giratorio. Dar por terminado el mezclado con dos o tres emboladas lentas y suaves. Sacar el émbolo inclinándolo ligeramente sobre la superficie de la suspensión para dejar caer las gotas de ésta adheridas. Anotar el tiempo inmediatamente. Añadir una gota de alcohol amílico si la superficie estuviese cubierta con espuma y mezclar de nuevo si fuese necesario. Una vez anotado el tiempo, la suspensión no se vuelve a mezclar.
Introducir el densímetro con cuidado en la suspensión, y después de treinta segundos a partir del tiempo anotado leer la escala siguiendo las instrucciones ya descritas y anotar la lectura. Sin sacar el densímetro hacer otra lectura cuando t = 60 segundos. Sacar entonces el densímetro con cuidado, enjuagarlo y secarlo con un paño. Introducir cuidadosamente el densímetro en la suspensión unos diez segundos antes de cada medida y hacer lecturas a los tres, diez, treinta y noventa minutos, y un tiempo superior a las cinco horas, que puede ser adoptado de conveniencia con la jornada laboral del laboratorio, anotando Cada vez la temperatura de la suspensión.
2(a).5. Cálculo
2.(a).5.1. Calcular para cada lectura la concentración de la suspensión expresada en g/l. y los porcentajes acumulados.
Concentración de la suspensión: C = L — L'.

L = lectura del densímetro.
L' = lectura de calibración del densímetro.
P' = peso seco del suelo en g/l. de la suspensión.
2(a).5.2 Calcular el tamaño correspondiente de las partículas o «diámetros», mediante la ecuación
x (μ) = Ө / (t)½
donde t es el tiempo de sedimentación en minutos y Ө se obtiene de la tabla 2(a).I en función de las lecturas observadas L (no corregidas).
Estos tamaños de las partículas han de corregirse cuando la temperatura es diferente de 30° C, multiplicándola por un factor «f» que se da en la tabla 2(a).II en función de la temperatura.
x' = f x

η = viscosidad del agua á la temperatura de sedimentación.
η30 = viscosidad del agua a 30° C.
2(a).5.3. Representar gráficamente en un sistema de coordenadas con escala semilogarítmica los valores de los diámetros de las partículas (x') en función de los valores de P obtenidos por 2(a).5.1, usando la escala logarítmica de abscisas para la representación del diámetro de las partículas (x') En la curva resultante se interpolan los porcentajes acumulados que se deseen para valores dados de x', p. e. 2, 5, 20 y 50.
Las fracciones de suelo menor de 2 mm. analizadas se denominan de la siguiente forma por el sistema internacional (Atterberg) y por el del Departamento de Agricultura de los Estados Unidos (U.S.D.A.)

En la figura 2(a).I se da el triángulo de clasificación de suelos por textura del U.S.D.A. Se recomienda usar la clasificación de familias que comprenden siete divisiones textuales. En esta división, la arena muy fina (0,10 a 0,05 mm.) es considerada como limo. Además, en los límites de las clases francas y limosas, los fragmentos gruesos se consideran equivalentes a la arena gruesa.
2(a).6. Observaciones
Conviene realizar esta determinación en un local a temperatura constante.
El grado de precisión de los resultados obtenidos con el densímetro depende entre otras cosas de la magnitud de la desviación de la densidad de las partículas del valor medio de 2,65 g/ml. Los porcentajes acumulados serán afectados en un ± 3 por 100 del valor medido dentro del normal intervalo de variación de la densidad de las partículas entre 2,5 y 2,8 g/ml. Este intervalo incluye la mayoría de los suelos, aunque los suelos tratados con H2O2 para destruir la materia orgánica pueden dar densidades ligeramente más altas. De manera que un porcentaje acumulado del 60 por 100 puede estar afectado de un error del ± 1,8 por 100 del peso seco de suelo dentro del intervalo de densidad mencionado.
Aunque el procedimiento de dispersión que se ha descrito es satisfactorio para muchos suelos, necesita ser modificado para algunas clases de suelos. Los siguientes tipos de suelos requerirán un tratamiento especial: suelos ricos en materia orgánica, suelos con yeso y suelos que contengan grandes cantidades de rocas o minerales blandos y fácilmente disgregables Cualquier soluto que se añada durante el pretratamiento, excepto el contenido en la solución dispersante, debe ser eliminado antes del análisis. Los suelos que contengan CO3Ca se dispersarán satisfactoriamente por el procedimiento descrito a menos que sean otros factores los que impidan la dispersión.
La magnitud de la dispersión es afectada por el tiempo de mezclado. Los cambios más rápidos ocurren durante los dos primeros minutos y generalmente alcanzan un máximo en menos de cinco minutos. Los fragmentos de rocas y minerales blandos pueden desintegrarse lentamente durante el mezclado, aumentando, por tanto, la cantidad de material fino presente en suspensión. Por esta razón, no es deseable prolongar el mezclado más allá del mínimo necesario para dispersar los componentes coloidales.
Las cuchillas de la batidora eléctrica se deterioran rápidamente por abrasión y deben de ser reemplazadas tan pronto como muestren signos de desgaste. A veces una o dos horas de operación son suficientes para provocar desgaste.
La agitación durante la noche en un agitador mecánico de tipo recíproco, tal como se describe en 2(b) puede ser un sustituto permisible del método de dispersión descrito. Sin embargo, deben usarse botellas de 500 ml. en vez de 250 ml., para evitar que el suelo se acumule en el fondo de las mismas.
2(a).7. Referencias
1. Day, P. R.: Particle Fractionation and Particle-size Analysis. «Methods of Soil Analysis». Part 1: 545-567. American Society of Agronomy, 1965.
2. Kilmer, V. J., y Alexander, L. T.: «Methods of making mechanical analysis of soil». Sci. 68: 15-24, 1949.
3. Soil Survey Staff. U.S.D.A., 1964. Draft. Subject to Change to «Soil Classification. A comprehensive System 7 th Approximation», 1960.
TABLA 2 (a).I.
Valores de Ө para la determinación de tamaño de las partículas en función de las lecturas observadas en el densímetro (Day, 1956)
| L | Ө | L | Ө | L | Ө |
|---|---|---|---|---|---|
| — 5 | 50.4 | ||||
| — 4 | 50.1 | 11 | 46.4 | 26 | 42.2 |
| — 3 | 49,9 | 12 | 46.2 | 27 | 41.9 |
| — 2 | 49.6 | 13 | 45.9 | 28 | 41.6 |
| — 1 | 49.4 | 14 | 45.6 | 29 | 41.3 |
| 0 | 49.2 | 15 | 45.3 | 30 | 41.0 |
| 1 | 48.9 | 16 | 45.0 | 31 | 40.7 |
| 2 | 48.7 | 17 | 44.8 | 32 | 40.4 |
| 3 | 48.4 | 18 | 44.5 | 33 | 40.1 |
| 4 | 48.2 | 19 | 44.2 | 34 | 39.8 |
| 5 | 47.9 | 20 | 43.9 | 35 | 39.5 |
| 6 | 47.7 | 21 | 43.7 | 36 | 39.2 |
| 7 | 47.4 | 22 | 43.4 | 37 | 38.0 |
| 8 | 47.2 | 23 | 43.1 | 38 | 30.6 |
| 9 | 47.0 | 24 | 42.8 | 39 | 38.3 |
| 10 | 46.7 | 25 | 42.5 | 40 | 38.0 |
TABLA 2(a).II.
Valores del factor de corrección f del diámetro de las partículas para temperaturas diferentes a 30° C
| t °C | f | t °C | f |
|---|---|---|---|
| 12 | 1.24 | 22 | 1.09 |
| 13 | 1.23 | 23 | 1.08 |
| 14 | 1.21 | 24 | 1.07 |
| 15 | 1.19 | 25 | 1.05 |
| 16 | 1.18 | 26 | 1.04 |
| 17 | 1.16 | 27 | 1.03 |
| 18 | 1.14 | 28 | 1.02 |
| 19 | 1.13 | 29 | 1.01 |
| 20 | 1.12 | 30 | 1.00 |
| 21 | 1.10 | 31 | 0.99 |
| 32 | 0.98 |
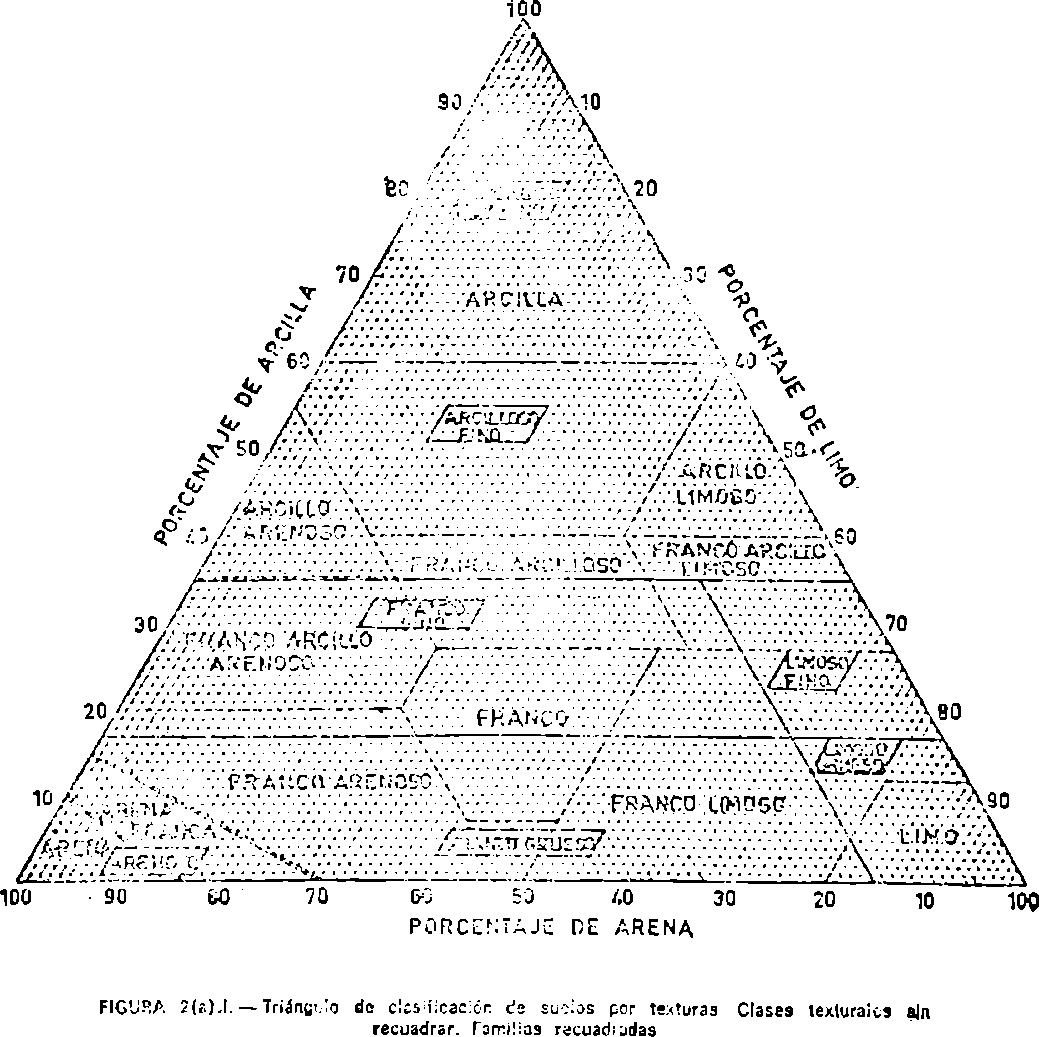
2(b) TEXTURA
(Método de la pipeta)
2(b).1. Principio
[Ver método 2(a).]
El método de la pipeta para el análisis de la textura está basado en el hecho de que la sedimentación elimina en una zona de profundidad h, y en un tiempo t, todas las partículas cuyas velocidades de sedimentación sean mayores de h/t, mientras que las partículas que tengan velocidades se sedimentación inferiores a la citada se mantienen en dicha zona a la concentración original. El muestreo de un pequeño volumen de la suspensión a la profundidad h después de un tiempo de sedimentación t proporciona una muestra en la que todas las partículas de diámetro mayor que x (según se determina por la ecuación de Stokes) han sido eliminadas, mientras que todas las partículas más finas están presentes en la muestra a la misma concentración que en la suspensión original. La muestra tomada a la profundidad «h» ha sido «tamizada» por sedimentación, de tal manera que la relación del peso «W» de las partículas presentes en la muestra después de un tiempo de sedimentación «t», dividido por el peso «Wo» de las partículas inicialmente presentes en el mismo volumen de la suspensión inicial, es igual a P/100, donde P es el porcentaje de partículas en peso, de diámetro menor que x. Ahora bien, la relación W/Wo puede también ser descrita como la relación de concentraciones c/Co, siendo c/Co = P/100. Esta ecuación relaciona la concentración c en la muestra, en g/l., con el parámetro P de distribución de partículas, siendo Co el peso de todas las partículas en toda la suspensión original dividido por el volumen de ésta.
2(b).2. Material y aparatos
2(b).2.1. Dispositivo de filtrado. Mediante un tubo de goma de paredes gruesas conectar un filtro de Pasteur-Chamberlan a un matraz Kitasato y a una pera de goma a través de una llave de paso con tres orificios, tal como muestra la figura 2(b).I. Conectar el Kitasato a la bomba de vacío.
2(b).2.2. Pipeta especial para toma de muestra. La pipeta se muestra en la figura 2(b).II. Determinar su capacidad a partir del peso de mercurio que sea capaz de alojar. Disponer también de un soporte de pipeta que permita bajar la posición de la misma a una profundidad controlada de 10 a 13 cm. por debajo de la marca superior de un litro de la probeta. Conectar la pipeta mediante un tubo de goma de 60 cm. de longitud a una línea de succión a través de un capilar y una llave de paso, como se muestra en la figura 2(b).III. Probar capilares diferentes hasta que se encuentre uno con el que la pipeta se llene bajo succión completa de treinta segundos.
2(b).2.3. Embolo agitador. Soldar en el centro de un disco de latón de 2 mm. de espesor y 50 mm. de diámetro una varilla de 500 mm. de longitud y 5 mm. de diámetro.
2(b).2.4. Agitador mecánico horizontal de tipo reciproco, con una elongación de 6 cm. y 120 vaivenes por minuto.
2(b).2.5. Juego de seis tamices con las siguientes aberturas de malla: 1,0; 0,5; 0,25; 0,177; 0,105; 0,047 mm.
2(b).2.6. Agitador vibrador de tamices.
2(b).3. Reactivos
2(b).3.1. Peróxido de hidrógeno (H2O2) al 30 por 100.
2(b).3.2. Solución de calgon [ver 2(a).3.1]. Disolver 50 g. de calgon en agua y diluir la solución en un litro.
2(b).4. Procedimiento
2(b).4.1. Pretratamiento. Pesar una muestra de unos 10 gramos, precisando hasta las centésimas, y colocarla en un vaso de pyrex de un litro al que se le añaden 30 ml. de agua. Tapar con un vidrio de reloj y mezclar bien removiendo el vaso. Añadir unos cuantos ml. de H2O2 al 30 por 100 y agitar la suspensión si es necesario para reducir la formación de espuma (tener cuidado de que el H2O2 no caiga sobre la piel) Cuando la reacción aminore, añadir cantidades adicionales de H2O2. Completar la digestión calentando el vaso durante una hora el menos a 90° C en un baño de agua o placa de calentamiento. Repetir el tratamiento hasta que se produzcan reacción da la suspensión con el agua oxigenada.
2(b).4.2. Filtración. Eliminar el exceso de líquido mediante el dispositivo de filtrado descrito, cuando la cápsula de filtro se haya recubierto de una capa de suelo de 1 ó 2 mm., invertir la llave de paso y ejercer presión con la pera de goma, haciendo que la cápsula de filtro roce ligeramente con la pared interior del vaso para hacer que el suelo se desprenda del filtro. Cuando el agua libre se haya eliminado, añadir más agua mezclando bien la suspensión. Filtrar y lavar varias veces, asegurándose en el último lavado que todo el suelo quede arrastrado en suspensión. Desecar el vaso con la suspensión en una estufa a 105° C, dejar enfriar en un desecador y pesar.
2(b).4.3. Dispersión y tamizado en húmedo. Añadir exactamente 10 ml. de la solución de calgon y remover el vaso para suspender las partículas (para comprobar la solución de calgon verter una alícuota de 10 ml. en un vaso tarado, secar en una estufa a 105° C durante una noche y pesar precisando hasta 0,001 g.). Transferir la suspensión a través de un embudo a frasco de 250 ml. y añadir agua hasta tener un volumen de suspensión de 180 ml., tapar bien y agitar el frasco en un agitador mecánico horizontal de tipo recíproco durante una noche. Colocar el frasco en posición vertical y dejar que se sedimente la suspensión durante unos minutos.
Colocar un embudo de boca ancha sobre una probeta de un litro. Comprobar que el tamiz de 47 μ esté limpio y sin roturas. Humedecerlo con agua por ambos lados y colocarlo, sobre el embudo. Verter, sin agitar ni remover, el frasco, la parte suspendida de la mezcla suelo-solución dispersante sobre el tamiz. Añadir agua al residuo que quede en el frasco, remover la mezcla y dejarla en reposo durante uno o dos minutos, decantando la porción suspendida en el tamiz. Repetir el proceso de mezclado y decantación varias veces hasta que la mayor parte del material fino haya sido transferido a la probeta. Poner boca abajo el frasco sobre el tamiz y arrastrar todas las partículas que permanezcan en el frasco con un chorro de agua de un frasco lavador. Cuando no quede ningún residuo en el frasco, proyectar un chorro de agua sobre el material retenido en el tamiz para obtener separación completa entre material fino y grueso. Añadir agua destilada al cilindro hasta completar un litro, colocar un tapón de goma (las pérdidas de agua por evaporación antes o durante la sedimentación pueden afectar los resultados) y colocar la probeta en el banco de sedimentación.
2(b).4.4. Tamizado en seco. Colocar el tamiz y la fracción por él retenida sobre una cápsula tarada y secar en una estufa. Enfriar y transferir la fracción arenosa a la cápsula con ayuda de una brocha suave. Volver a secar la cápsula durante dos horas a 105° C, enfriar en un desecador y pesar la cápsula y la arena precisando hasta 0,01 g.
Comprobar que el juego de tamices esté limpio y en buenas condiciones mecánicas. Los de malla más fina son particularmente susceptibles a la extensión de los hilos y rotura de la tela mecánica en los bordes. Colocar los tamices sobre la batea en el siguiente orden, de arriba abajo: 1.000, 500, 250, 177, 105 y 47 μ. Transferir la fracción arenosa de la cápsula al tamiz superior y tamizar.
Colocar la fracción retenida por cada tamiz en la cápsula tarada, empezando por el tamiz más grueso. Pesar la cápsula y los sucesivos pesos acumulados después de cada adición. Incluir el peso del «limo residual» que haya caído sobre la batea inferior, ya que tal peso es parte de la fracción limosa gruesa. Comparar el peso acumulado de todas las fracciones con el peso total de las fracciones gruesas previamente determinado.
2(b).4.5. Sedimentación y muestreo con pipeta. Anotar la temperatura de la suspensión cuando se estabilice. Sumergir el émbolo en la suspensión y moverlo verticalmente para mezclar bien. Sostener la probeta con firmeza cuando se tire del émbolo hacia arriba. El movimiento de éste en las proximidades de la superficie libre debe de ser suave para no derramar el contenido. Dar emboladas fuertes cuando se tire del émbolo en las proximidades del fondo para arrastrar cualquier partícula que haya podido depositarse en él. Desalojar los sedimentos depositados en los bordes inferiores del fondo del cilindro, inclinando la varilla del agitador e imprimiéndole un movimiento giratorio. Dar por terminado el mezclado con dos o tres emboladas lentas y suaves, inclinando el émbolo por encima de la suspensión al sacarlo para dejar caer las gotas de ésta adheridas. Anotar el tiempo inmediatamente.
Colocar la probeta junto a la pipeta lista para succión. Previamente se ha hecho el ajuste preciso para sumergir la pipeta 10 cm. en la suspensión. Anotar el volumen de la pipeta en cuestión y determinar a partir de la tabla 2(b).I el tiempo de sedimentación requerido para el muestreo de las partículas mayores de 20 μ. Sumergir la pipeta a la profundidad descrita treinta segundos antes del tiempo de muestreo.
Abrir la llave de succión al tiempo de muestreo y cerrarla en el instante que se llene la pipeta. Subir la pipeta, secarla con un paño suave y abrir la llave para que se vierta la muestra de suspensión sobre una cápsula previamente tarada (hasta 0,001 g.). Enjuagar la pipeta y verter el líquido de lavado sobre la cápsula que se coloca en una estufa a 105° C durante doce horas. Enfriar en un desecador y pesarla precisando hasta 0,001 g.
Tomar muestras de 5 y/o 2 μ de la misma manera, volviendo a agitar la suspensión o no, según se desee, pero siempre ha de contarse el tiempo desde el instante del último agitado. Medir siempre la profundidad de muestreo desde la superficie de la suspensión y no desde el nivel usado para el muestreo precedente.
2(b).5. Cálculo
Calcular el peso aparente de las partículas en el intervalo de tamaño dado por la fórmula

P = peso seco de la muestra de la pipeta.
V = volumen total de la suspensión.
V' = volumen de la pipeta.
Deducir el peso del agente dispersante por litro de suspensión, usando los datos obtenidos por desecación de una muestra de 10 ml. del reactivo 0,5 N. Este resultado será el peso acumulado ΣP.
Determinar por diferencia los pesos P de las fracciones individuales, tal como se describe en el ejemplo de la tabla 2(b).II. Determinar los pesos de las fracciones de arena tal como se hace en tabla 2(b).III.
Calcular las pérdidas de solución y de tamizado tal como se hace en la tabla 2(b).IV. El término de pérdidas de solución se refiere no sólo a los solutos eliminados por lixiviación, sino también a las pérdidas de materia orgánica.
Calcular el peso del limo grueso.
Resumir los datos de las tablas 2 (b).II, III y IV en una tabla [tal como se hace en la tabla 2(b).V].
La denominación de las fracciones del suelo menor de 2 milímetros se hace de acuerdo con la terminología dada en 2(a).4. La clasificación de textura se hace de acuerdo con el diagrama de la figura 2(a).I.
2(b).6. Observaciones
Esta determinación debe realizarse en local a temperatura constante.
2(b).7. Referencia
1. Day, P. R.: «Particle Fractionation and Particle-size Analysis», «Methods of Soils Analysis», part 1: 545-567. American Society of Agronomy, 1965.
TABLA 2(b).I
Tiempo de sedimentación para partículas de 2, 5 y 20 μ de diámetro en agua para una profundidad de 10 centímetros
| Tiempo de sedimentación para los diámetros indicados | ||||||
|---|---|---|---|---|---|---|
|
Temperatura °C |
2 MICRAS | 5 MICRAS | 20 MICRAS | |||
| horas | minutos | horas | minutos | minutos | segundos | |
| 20 | 8 | 0 | 1 | 17 | 4 | 48 |
| 21 | 7 | 49 | 1 | 15 | 4 | 41 |
| 22 | 7 | 38 | 1 | 13 | 4 | 35 |
| 23 | 7 | 27 | 1 | 11 | 4 | 28 |
| 24 | 7 | 17 | 1 | 10 | 4 | 22 |
| 25 | 7 | 7 | 1 | 8 | 4 | 16 |
| 26 | 6 | 57 | 1 | 7 | 4 | 10 |
| 27 | 6 | 48 | 1 | 5 | 4 | 4 |
| 28 | 6 | 39 | 1 | 4 | 4 | 0 |
| 29 | 6 | 31 | 1 | 3 | 3 | 55 |
| 30 | 6 | 22 | 1 | 1 | 3 | 49 |
| 31 | 6 | 14 | 1 | 0 | 3 | 44 |
TABLA 2(b).II
Cálculos de la muestra de la pipeta
| Tamaño partícula mm. | Peso muestra g. | Volumen muestra ml. | Volumen suspensión ml. | PV/V' g. | ∑P | P |
|---|---|---|---|---|---|---|
| 0,020 | 0,114 | 26,49 | 1.000 | 4,30 | 3,81 | 1,54 |
| 0,005 | 0,073 | 26,49 | 1.000 | 2,76 | 2,27 | 0,61 |
| 0,002 | 0,057 | 26,49 | 1.000 | 2,15 | 1,66 | 1,66 |
TABLA 2 (b).III
Cálculos de la muestra del tamizado
| Tamaño nominal mm. | Peso de arena g. | Peso de las fracciones g. |
|---|---|---|
| 2 | 0,00 | 0,00 |
| 1 | 0,01 | 0,01 |
| 0,5 | 0,03 | 0,02 |
| 0,25 | 0,06 | 0,03 |
| 0,10 | 0,58 | 0,52 |
| 0,05 | 2,02 | 1,44 |
| 0,00 | 2,31 | 0,29 |
TABLA 2(b).IV
Para muestras de suelos alteradas
Cálculo de la muestra total
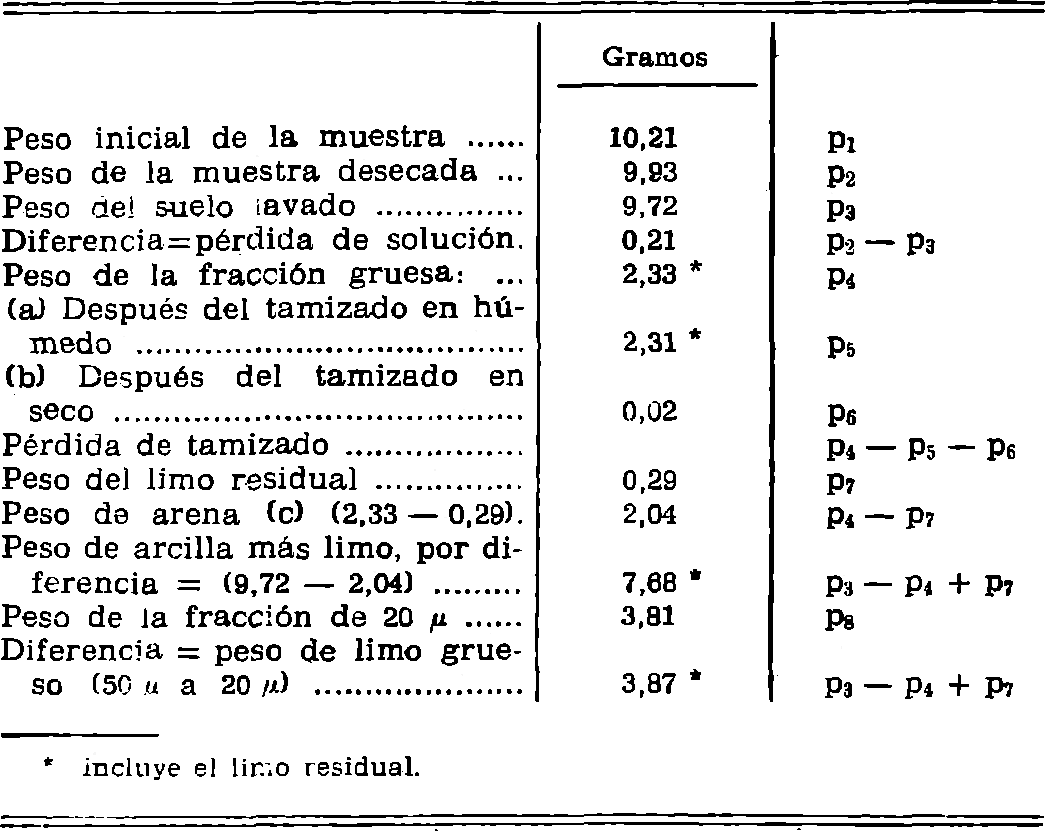
TABLA 2(b).V
Resumen de los cálculos
| Tamaño nominal mm. | Peso fracción g. | p* % | p** % |
|---|---|---|---|
| 2-1 | 0,01 | 0,1 | 99,8 (a) |
| 1-0,5 | 0,02 | 0,2 | 99,7 |
| 0,5-0,25 | 0,03 | 0,3 | 99,5 |
| 0,25-0,1 | 0,52 | 5,4 | 99,2 |
| 0,1-0,05 | 1,44 | 14,8 | 93,8 |
| 0,05-0,02 | 3,87 | 39,8 | 79,0 |
| 0,02-0,005 | 1.54 | 15,8 | 39,2 |
| 0,005-0,002 | 0,61 | 6,3 | 23,4 |
| 0,002-0 | 1,66 | 17,1 | 17,1 |
|
p* = (peso de la fracción) (100/peso del suelo lavado). p** = porcentaje de partículas en peso, más pequeñas que el límite superior de la primera columna. (a) = pérdidas por tamizado 0,2 por 100. |
|||
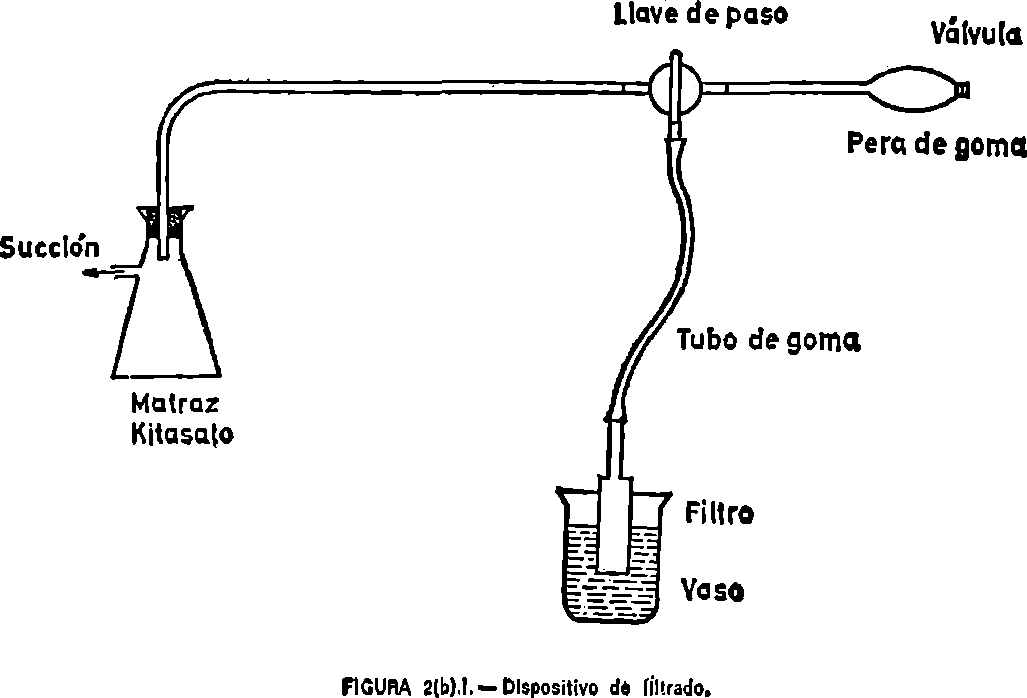
3(a) CONDUCTIVIDAD HIDRAULICA
(Columnas de suelo inalteradas y saturadas)
3(a).1. Principio
La conductividad hidráulica del suelo se define con el coeficiente de proporcionalidad de la ley de Darcy, aplicada al flujo de agua en el suelo. Esto es:
V = K i (1)
en donde V es la velocidad del flujo;
i es el gradiente hidráulico;
K es la conductividad hidráulica.
V puede expresarse como el volumen (V') de agua que fluye a través de un área determinada (A) en cierto tiempo (t). Entonces:

i es por definición la pérdida de carga (h) dividido por la distancia (l).
 (2)
(2)
La permeabilidad del suelo en un sentido cuantitativo o permeabilidad intrínseca (K') se define, sin referencias a un fluido en particular, como la constante de proporcionalidad en la siguiente expresión general de la ley de Darcy.
 (3)
(3)
en donde d y η son la densidad y viscosidad del fluido, respectivamente, y g es la aceleración de la gravedad. Las dimensiones de K' son las de un área.
3(a).2. Material y aparatos
3(a).2.1. Equipo de toma de muestras inalteradas.
3(a).2.2. Depósito de agua a nivel constante y grada soporte de las muestras [figura 3(a).I].
3(a).2.3. Probetas de 100 ml.
3(a).3. Procedimiento
Colocar los cilindros con las muestras en la batea de saturación tal como se muestra en la figura 3(a).1. Dejar en remojo durante unas veinte horas para que se saturen. Por este procedimiento de saturación por ascenso capilar del agua, los cambios estructurales que la expulsión rápida del aire puede acarrear son mínimos. A continuación adosar un collarín suplementario a un extremo de cada cilindro sujetado con una banda ancha de goma o cinta adhesiva impermeable al agua, manteniendo el otro extremo del cilindro en contacto con el agua. Verter agua poco a poco sobre el cilindro hasta que alcance un nivel en el suplemento de 2/3 ó 3/4 del total. Transferir rápidamente las muestras a la grada de percolación colocando cada cilindro sobre un papel de filtro ajustado a un tamiz metálico (2 mm. de abertura) y conectarlos por medio de un sifón al depósito de agua a nivel constante. No dejar que el agua rebose por el borde superior del collarín.
Después de que el nivel de agua se haya estabilizado, recoger el agua que percole en probetas de 100 ml. Medir el volumen de agua V que percole en un tiempo conocido t. Medir la pérdida de carga h.
3(a).4. Cálculo
Calcular la conductividad hidráulica mediante la fórmula

Expresar V en ml., l y h en las mismas unidades, A en cm2 y t en horas, en lo que la conductividad hidráulica vendrá dada en cm/hora.
3(a).5. Observaciones
El hecho de que la conductividad hidráulica sea el factor de proporcionalidad de la ley de Darcy no significa que sea o deba ser una constante. Debido a procesos químicos, físicos y biológicos que ocurren en el suelo se pueden producir cambios de la conductividad conforme el agua pasa a través de la columna de suelo [3(a).6.2].
La conductividad hidráulica es particularmente sensible al cambio de los cationes de saturación del suelo. La conductividad tiende a ser directamente proporcional a la concentración de iones en el agua que percole. Por ello, debe de usarse agua de la misma calidad que la que exista en el campo (agua de riego en caso de un suelo que se riegue o vaya a regar). Si esto no es posible no debe usarse agua destilada, sino agua corriente.
En la tabla 3(a).I se dan las clases de permeabilidad y los valores correspondientes de conductividad hidráulica para muestras de suelo saturadas
Es necesario siempre duplicar las determinaciones. En la tabla 3(a).II se dan los límites de confianza de la conductividad hidráulica en función del número de muestras y del valor medio de la conductividad.
3(a).6. Referencias
1. Klute, A.: «Methods of Soil Analysis». Part 1: 210-221. American Society of Agronomy, 1965.
2. Reeve, R. C.: «Factors which affect permeability». Agron J. 7: 404-414, 1957.
3. United States Salinity Laboratory Staff: «Diagnosis and Improvement of Saline and Alkali Soils», 1954.
TABLA 3(a).I
Clases de permeabilidad de suelos saturados e intervalos correspondientes de la conductividad hidráulica y permeabilidad
| Clase | Conductividad hidráulica cm/hora | Permeabilidad cm2 ( x 10‒10) |
|---|---|---|
| Muy lenta. | < 0,125 | < 3 |
| Lenta. | 0,125 - 0,5 | 3 - 15 |
| Moderadamente lenta. | 0,5 - 2,0 | 15 - 60 |
| Moderada. | 2,0 - 6,25 | 60 - 170 |
| Moderadamente rápida. | 6,25 - 12,5 | 170 - 350 |
| Rápida. | 12,5 - 25,0 | 350 - 700 |
| Muy rápida. | >25,0 | >700 |
TABLA 3(a).II
Limites de confianza de la conductividad hidráulica en función del número de muestras y de la conductividad media
| Número de muestras | Límite de confianza K |
|---|---|
| 2 | 2,00 K |
| 3 | 1,90 K |
| 5 | 1,45 K |
| 8 | 1,11 K |
| 16 | 0,76 K |
3(b) CONDUCTIVIDAD HIDRAULICA
3(b).1. Material y aparatos
El cilindro en donde se coloca la muestra de suelo se construye cortando tubo de latón de 7,5 y 1,25 cm, de diámetro exterior y 1,5 a 2 mm. de espesor y cortando de una plancha de latón de igual espesor un disco de 7,5 cm. de diámetro, en cuyo centro se perfora un orificio de 1,25 cm. de diámetro. Las dimensiones se dan en la figura 3(b).I y las partes se sueldan con estaño (o plomo y estaño).
En el fondo del cilindro se coloca un tamiz recortado de una malla de bronce de orificios redondos y 2 mm. de abertura.
Para la compactación de la muestra se construye un bloque de madera como el representado en la figura 3(b).II. La perforación central es para que se adapte con holgura el tubo de salida del agua y las varillas para asegurar la caída vertical del cilindro durante el proceso de compactación. La varilla que sobresale 2,5 mm. por encima del cilindro en reposo sirve como referencia de la altura de caída.
Se requiere, además, como en el método 3(a), un depósito de agua a nivel constante, grada soporte de las muestras y probetas de 100 ml.
3(b).2. Procedimiento
Colocar un papel de filtro de tamaño adecuado sobre el tamiz del cilindro.
Transferir una muestra de 200 g. de suelo desecado al aire y verterla con un movimiento brusco en el cilindro de percolación. Así se impedirá la segregación de partículas de diferentes tamaños.
Dejar caer veinte veces el cilindro sobre el bloque de madera desde una altura de 2,5 cm. Colocar un papel de filtro en la superficie del suelo y verter poco a poco agua en el cilindro de manera que se altere lo menos posible la muestra compactada. Anotar la hora en la que se añade el agua y, si es posible, la hora a que comience a salir. Recoger el agua percolada en una probeta y medir el volumen recogido en intervalos de tiempo conveniente. Las mediciones se continúan hasta que el agua que haya percolado a través de la muestra equivalga a una altura de agua de 12 cm. sobre la superficie del suelo. En los suelos de percolación muy baja se debe de intentar obtener al menos una medición del flujo, siendo el tiempo más bien que la altura de agua percolada el factor a tener en cuenta para dar por finalizada la percolación.
3(b).3. Cálculo
Calcular la conductividad hidráulica (K) aplicando la fórmula
V = volumen de agua en ml. que percola a través de la columna de suelo en el tiempo t (horas).
A = área de la sección de la columna del suelo en cm2.
l = altura de la columna de suelo expresada en las mismas unidades que la pérdida de carga h.
La pérdida de carga debe de medirse desde el nivel constante de la superficie del agua sobre el suelo hasta la base de la columna por donde desagua la muestra (tensión del agua nula).
La altura de la columna de suelo debe de medirse durante o después de haberse percolado el agua y no cuando el suelo esté seco.
3(b).4. Observaciones
Aunque teóricamente ni el diámetro ni la altura de la columna de suelo deben de afectar a las mediciones, los resultados más consistentes se obtienen cuando la altura de la columna es menor que el diámetro, especialmente con muestras de suelos que se hinchan al humedecerse. La experiencia aconseja que el cilindro que contenga la muestra debe de tener al menos 7,5 cm. de diámetro para una profundidad del suelo de 5 cm.
Las medidas de conductividad hidráulica deben de ser hechas en el intervalo de temperatura 18° a 24° C. El efecto de la temperatura en este intervalo sobre la conductividad hidráulica es pequeño comparado con otros factores, tales como los derivados de la calidad del agua, cationes cambiables y salinidad del suelo.
El gradiente hidráulico (h/l) suele fijarse entre 1 y 4, aunque valores del orden de 10 no parecen afectar los resultados.
En general, el mismo tipo de agua que se vaya a utilizar para el riego debe de usarse en el laboratorio para la determinación de la conductividad hidráulica.
Normalmente deben hacerse determinaciones por triplicado. En el caso que el intervalo de variación exceda el 50 por 100 del valor medio de la conductividad hidráulica se repiten las determinaciones con otras muestras, desestimando la primera serie de valores. Las diferencias entre valores de conductividad hidráulica del orden del 15 por 100 al 20 por 100 entre distintos suelos o tratamientos no se consideran significativas.
3(b).5. Referencia
1. United States Salinity Laboratory Staff: «Diagnosis and Improvement of Saline and Alkali Soils», pp. 112-113, 1954.
4. DENSIDAD APARENTE
4.1. Principio
La densidad aparente (Da) del suelo es la relación de la masa al volumen macroscópico ocupado por las partículas del suelo más el espacio poroso. La masa se determina pesando la muestra desecada a 105° C y el volumen de la muestra que se haya tomado en el campo.
4.2. Material y aparatos
Equipo de toma de muestras inalteradas. En su defecto puede usarse un trozo de tubo de pared delgada con bordes cortantes en el extremo que se presione en el suelo.
4.3. Procedimiento
Enrasar con espátula o navaja el suelo que sobresalga del cilindro muestrador. El volumen del suelo se calcula de las dimensiones interiores de éste. En algunos modelos, el filo cortante del cilindro tiene un diámetro ligeramente inferior al del resto del cilindro para así reducir la fricción que se produce al entrar el suelo en el cilindro. En este caso, se utilizará el diámetro del borde cortante del cilindro para calcular el volumen del suelo. Transferir el suelo a un recipiente para determinación del peso seco como en 1.
4.4. Cálculo
Expresar la densidad aparente en gramos de suelo seco por centímetro cúbico.
4.5. Observaciones
Aunque la determinación de la densidad aparente no requiere que las muestras se mantengan en los cilindros para su transporte del campo al laboratorio, será conveniente mantenerlas inalteradas y conservar su contenido en agua cuando se quiere determinar la humedad de las mismas. Para ello los cilindros muestreadores pueden meterse en cajas cilíndricas de cartón encerado o en latas con tapaderas para evitar la pérdida de agua durante el transporte.
5. DENSIDAD REAL
5.1. Principio
El término densidad real del suelo se refiere a la densidad de las partículas sólidas estimada colectivamente. Se expresa como la relación de la masa total de las partículas sólidas a su volumen total, excluyendo el volumen ocupado por los poros entre las partículas.
La masa de las partículas se determina pesándolas y su volumen se calcula a partir de la masa y densidad del agua u otro fluido que es desplazado por la muestra de suelo.
5.2. Material y aparatos
Picnómetro. Si no se dispone de picnómetro puede usarse un matraz aforado de 25, 50 ó 100 ml., siempre que se opere con una muestra lo suficientemente grande como para compensar la menor precisión al medir el volumen del líquido.
5.3. Procedimiento
Pesar el picnómetro limpio y seco al aire. Añadir 10 g. de suelo secado al aire. En caso de utilizar un matraz aforado de 100 ml. añadir 50 g. de suelo. Limpiar el exterior y el cuello del picnómetro si se ha impregnado de suelo durante el transvase. Pesar el picnómetro incluyendo el tapón. Determinar la humedad de una muestra duplicada de suelo desecando en estufa a 105° C como en 1.
Llenar el picnómetro hasta la mitad con agua destilada arrastrando hacia el interior las partículas de suelo que se hayan podido adherir al cuello. Expulsar el aire ocluido hirviendo suavemente el agua durante algunos minutos y agitando el contenido suavemente para evitar pérdida de suelo por formación de espuma.
Enfriar a temperatura ambiente y añadir agua destilada y hervida hasta llenar el picnómetro. Tapar, secar y limpiar el exterior con un paño seco. Pesar de nuevo y anotar la temperatura del agua en el picnómetro.
Finalmente, sacar el suelo del picnómetro y llenarlo con agua destilada y hervida que esté a la misma temperatura que la anterior. Tapar, limpiar y secar como antes y pesar, anotando la temperatura del agita.
5.4. Cálculo
Calcular la densidad real (Dr) por la siguiente fórmula:

da = densidad del agua en g/ml. a la temperatura observada.
Ps = peso del picnómetro + muestra de suelo.
P'a = peso del picnómetro lleno de aire.
Psa = peso del picnómetro lleno de suelo + agua.
Pa = peso del picnómetro lleno de agua a la temperatura observada.
Este método es muy preciso cuando se efectúan las pesadas con precisión. Un error de 2 mg. en una muestra de 10 g. de suelo origina un error en la densidad real del orden de 0,0003 g/ml. Un error de 10 mg. en una muestra de 30 g. de suelo conduce a un error de 0,001 g/ml.
6. PORCENTAJE DE SATURACION
6.1. Material y aparatos
Como en 1,2.
6.2. Procedimiento
Transferir una porción de la pasta saturada del suelo preparada según el método 13(a) (Métodos químicos) a un recipiente previamente tarado con tapadera. Determinar la humedad de la pasta según el método 1.
6.3. Cálculo

P' = pérdida de peso en g. por desecación.
P = peso en g. de suelo seco.
7. PORCENTAJE A 1/10 DE ATMOSFERA
7.1. Principio
Las muestras de suelo saturadas de agua se colocan en una cámara de presión sobre una membrana porosa en contacto con su cara inferior con agua libre a la presión atmosférica. Mediante la aplicación de una sobrepresión en la cámara se equilibra el suelo con el agua que es retenida a una cierta succión métrica. La membrana debe ser permeable al agua y a los solutos, pero no a la matriz del suelo.
7.2. Material y aparatos
7.2.1. Equipo de presión: membrana cerámica y cámara de presión (1).
(1) Existen en el mercado equipos especiales fabricados para estas determinaciones (métodos 7, 8 y 9), con membranas de 28 cm, de diámetro y que cubren el intervalo de presión de 0 a 1 atm. ó 0 a 15 atm.
7.2.2. Anillos de goma retenedores de las muestras del suelo de, aproximadamente, 1 cm. de altura y 6 cm. de diámetro, que pueden alojar al menos 25 g. de suelo.
7.2.3. Fuente de aire comprimido con presión regulable.
7.2.(4, 5 y 6). Como 1.2. (1, 2 y 3).
7.3. Procedimiento
Una vez que se haya comprobado el buen funcionamiento de la membrana porosa (ver 7.5) tomar muestras por duplicado de 25 g. de suelo tamizado por una malla de 2 mm. Colocar los anillos retenedores de la muestra sobre la membrana porosa. Para evitar segregación de partículas por tamaño, vaciar todo el suelo en el anillo de una vez y nivelar. Dejar las muestras en reposo durante al menos dieciséis horas con un exceso de agua en la membrana. Tapar la cámara y aplicar una presión de 0,1 Kg/cm2. Las muestras de 1 cm. de altura pueden retirarse después de cuarenta y ocho horas de haberse iniciado la extracción, o bien cuando las lecturas en una bureta conectada al tubo de salida de agua de la membrana indiquen que ha cesado el flujo de agua. Algunos suelos requieren para aproximarse a su equilibrio de dieciocho a veinte horas. Antes de quitar la presión en la cámara es conveniente poner una pinza que cierre el tubo de salida de agua de la membrana. Esto evita movimiento de agua hacia las muestras al quitar la presión. Para evitar cambio de humedad de las muestras, colocarlas inmediatamente en los recipientes para determinación de humedad según se describe en el método 1.
7.4. Cálculos
Expresar la humedad en tanto por ciento de agua referido a suelo seco a 105° C.
7.5. Observaciones
Para comprobar posibles defectos de la membrana de presión que se vaya a usar deben de colocarse en la cámara de presión. Llenar la cámara de agua, taparla y aplicar una presión de 1 Kg/cm2. El flujo de agua hacia el exterior debe ser de 1 cm3/cm2 por hora y por atmósfera de gradiente de presión o mayor para asegurar un buen funcionamiento del equipo. A continuación, comprobar el reflujo de agua hacia el interior de la membrana de la siguiente forma: quitar la presión aplicada y vaciar el exceso de agua de la cámara y de las membranas, cerrar la cámara y aplicar una presión de 1/2 atm. o cualquier otra inferior al máximo que se vaya a usar en medidas de retención. Después de unos minutos, el flujo de agua hacia el exterior debe de haber cesado, y en condiciones ideales no deben de observarse burbujas de aire. En realidad puede tolerarse algún burbujeo del orden de 2 ó 3 cm3 de aire/minuto. Después de terminar esta prueba comprobar que no haya escapes por la tapa o cualquier otra conexión, sumergiendo la cámara en agua, o bien observar el cambio de presión en la cámara una vez desconectada del tanque de aire comprimido, para asegurar que la cámara y la membrana son impermeables al aire. Los escapes de aire pueden producir errores en las determinaciones de la humedad retenida a diversas succiones, debido a modificaciones de las muestras de suelos equilibrados que podrían perder agua por evaporación.
7.6. Referencia
1. United States Salinity Laboratory Staff: «Diagnosis and Improvement of Saline and Alkali Soils», p. 109, 1954.
8. PORCENTAJE A 1/3 DE ATMOSFERA
8. 1. Material y aparatos
Como en 7.
8.2. Procedimiento
Como en 7.4, excepto que la presión aplicada debe de ser de 0,34 Kg/cm2.
9. PORCENTAJE A 15 ATMOSFERAS
9.1. Material y aparatos
9.1.1. Equipo de presión con membrana para extracciones, a 15 atmósferas y cámara de presión.
9.1.(2, 3 y 4). Como 7.2 (2, 3 y 4) (1).
(1) Ver nota (1) método 7.
9.2. Procedimiento
Preparar muestras duplicadas de 25 g. de suelo tamizado por una malla de 2 mm. Humedecer la membrana e instalarla en el aparato, recortándola con una navaja alrededor del disco de bronce. Colocar los anillos retenedores de muestra sobre la membrana. Para evitar segregación de la muestra por tamaño de las partículas verter toda la muestra de un golpe en el anillo de retención. Si sólo se vierte una parte de muestra no seria representativa. Nivelar el suelo en el anillo, cubrir con un papel encerado y dejar en reposo con un exceso de agua durante dieciséis horas. Eliminar el exceso de agua de la membrana con una pipeta o una pera de goma. Cerrar la cámara de presión y dejar entrar aire a una presión de 15 atmósferas.
Después de unas cuantas horas, hay un descenso notable de la cantidad de agua que fluye hacia fuera de la membrana, lo cual se produce principalmente por la baja conductividad hidráulica del suelo y no por la permeabilidad de la membrana. En este momento las muestras de suelo tienen suficiente rigidez y compactación, por lo que se puede aplicar una presión diferencial de 0,28 Kg/cm2 sobre el diafragma de goma de la parte superior de la cámara. Esta acción del diafragma mantiene la muestra en contacto con la membrana, acelerando la extracción del agua en suelos de textura fina que se contraen fácilmente. El uso del diafragma no es necesario para suelos de textura media o gruesa.
Sacar las muestras en cualquier momento después de que hayan pasado cuarenta y ocho horas del comienzo, de la extracción o cuando las lecturas de una bureta conectada al tubo de salida de agua de la membrana indiquen que el flujo ha cesado: La mayoría de los suelos alcanzarán un equilibrio hidráulico con la membrana a las dieciocho o veinte horas de aplicar la presión, pero algunos requerirán un tiempo considerablemente más largo. Para evitar cambios de humedad de las muestras, transferirlas a los recipientes de humedad y determinar ésta según se indica en el método 1.
9.3. Cálculo
Expresar la humedad en tanto por ciento de agua a 105° C.
9.4. Observaciones
Debe tenerse cuidado de no poner el suelo pegado a la empaquetadura inferior de la cámara. El flujo que ocurre a través de la empaquetadura cuando la cámara está cerrada puede presionar las partículas de arena sobre la membrana y causar roturas en la misma.
9.5. Referencia
1. United States Salinity Laboratory Staff: «Diagnosis and Improvement of Saline and Alkali Soils», pp. 109-110, 1954.
10. AGREGACION DE PARTICULAS INFERIORES A 50 MICRAS
10.1. Principio
Se trata de un método rápido para estimar la estabilidad en agua de los agregados en los que intervienen partículas de la fracción arcilla y limo (menores de 50 μ). Este método requiere la medida de la concentración de dos suspensiones del mismo suelo, una de las cuales se dispersa por el procedimiento de dispersión convencional ya descrito en el método 2(a) para obtener el limo y la arcilla totales. En la otra suspensión, preparada por agitación suave de la muestra, se mide la cantidad de limo y arcilla no agregados. La diferencia de concentración entre las dos suspensiones proporciona una medida de la cantidad de partículas de limo y arcilla unidas en agregados estables al agua de tamaño superior a 50 μ.
Este procedimiento puede utilizarse también como una prueba rápida exploratoria para determinar el efecto de productos con poder agregante en la formación de agregados estables al agua.
10.2. Material y aparatos
10.2.(1, 2, 3 y 4). Como 2(a),2.(1, 2, 3 y 4).
10.3. Procedimiento
10.3.1. Muestreo. Tomar con pala la muestra del suelo preferentemente cuando esté húmedo, evitando la compactación o fragmentación excesiva del suelo. Desecar la muestra lentamente, y cuando esté suficientemente friable, tamizarla suavemente a través de un tamiz de 8 mm. y desecar al aire. Si el suelo es pedregoso, pasar la muestra a través de un tamiz de 4 mm. y separar todo el material de tamaño superior a 4 mm.
10.3.2. Súbmuestreo. Pesar, con aproximación de 0,1 g., dos submuestras de 50 g.
10.3.3. Dispersión. Colocar una de las submuestras en el vaso de la batidora y añadir unos 400 ml. de agua destilada.
Dispersar y transferir a la probeta completando con agua hasta un volumen total de 1.130 ml. Tapar y agitar la probeta invirtiéndola enérgicamente, sobre cada una de sus bases.
10.3.4. Preparación de la muestra no dispersada. Colocar otra de las submuestras en una probeta e inclinarla en una posición casi horizontal y agitar suavemente para extender la muestra en una distancia de 10 a 12 cm. a lo largo de la pared de la probeta. Añadir agua destilada poco a poco de tal manera que se favorezca el mojado del suelo por capilaridad mejor que por inundación. Cuando el suelo esté completamente mojado, añadir agua hasta completar un volumen total de la suspensión de 1.130 ml., pero sin dejar que el agua caiga directamente sobre el suelo. Dejar que el suelo se remoje durante unos quince minutos como mínimo. Tapar la probeta e invertir veinte veces suavemente (sin agitar) durante un período de cuarenta segundos, empleando un segundo entre inversiones.
10.3.5. Medida de la concentración de las suspensiones. Introducir el hidrómetro en la suspensión unos diez segundos antes del tiempo fijado para la lectura. Hacer la lectura a los cuarenta segundos de haber terminado la agitación o mezclado de las suspensiones en la probeta. Anotar inmediatamente la temperatura de la suspensión y corregir la lectura del hidrómetro, aumentándola o disminuyéndola en 0,11 g. por cada grado de más o menos, respectivamente, sobre la temperatura de 19,4° C.
La lectura corregida da el limo y arcilla en suspensión expresados en g/l.
10.4. Cálculo

C = concentración en g/l. (limo + arcilla totales) en suspensión dispersada.
C' = concentración en g/l. (limo + arcilla dispersados) en suspensión sin dispersar.
10.5. Referencia
1. United States Salinity Laboratory Staff: «Diagnosis and Improvement of Saline and Alkali Soils», pp. 125-126, 1954.
11. COEFICIENTE HIGROSCOPICO
11.1. Principio
Se fija vapor de agua sobre una muestra de suelo, procedentes de una atmósfera confinada cuya humedad relativa es del 90 por 100, según el Bureau of Soils (Washington) y A. Demolon, pesando el agua fijada. No se emplea la atmósfera saturada de humedad, para evitar condensaciones directas posibles al descender la temperatura.
11.2. Material y aparatos
11.2.1. Bomba de vacío.
11.2.2. Desecador con tubuladora de vacío.
11.2.3. Manómetro pequeño de mercurio (truncado).
11.2.4. Pesasustancias de forma baja.
11.3. Reactivos
11.3.1. Acido sulfúrico diluido (2 por 100 en volumen o 3,3 por 100 en peso).
11.4. Procedimiento
Poner unos 5 g. del suelo en un pesasustancias, previamente tarado con su tapa formando capa fina. Colocar el pesasustancias destapado en el desecador que contiene ácido sulfúrico diluido e introducir también dentro del desecador el manómetro de mercurio. Tapar el desecador, cuidando de dar vaselina a la tapa para que el cierre sea hermético. Reducir mediante vacío la presión en el interior del desecador a 30 mm. y mantener el desecador durante cinco días a una temperatura próxima a 25° C. Transcurrido este tiempo, extraer el pesasustancias destapado en una estufa a 110° C y desecar hasta peso constante. Deducir la humedad fijada por la muestra de suelo desecado y referirla a 100 g. de suelo desecado.
11.5. Referencias
1. Demolon, A.: Leroux, D.: «Guide pour l’étude experimentalé du sol». París, 1952.
2. Robinson, G. W.: «Los suelos». Barcelona, 1960.
12. INDICE DE INESTABILIDAD ESTRUCTURAL
12.1. Principio
La estabilidad de un agregado en la solución del suelo depende, entre otros factores, de la concentración de sales en dicha solución, aumentando con dicha concentración. Por otra parte, la destrucción de los agregados del suelo y subsecuente dispersión de los coloides aumenta el volumen del suelo. Comparando el volumen de sedimentación de muestras replicadas de suelo en agua destilada y en solución salina se puede estimar la estabilidad de los agregados del suelo.
12.2. Material y aparatos
12.2.1. Probetas de 50 cc. graduadas en décimas.
12.2.2. Varillas de alambre de 15 cm. de longitud.
12.3. Reactivos
Solución de cloruro potásico 1 N. Disolver 74,56 g. de cloruro potásico en agua y diluir en un litro.
12.4. Procedimiento
Tomar las probetas y llenarlas hasta 40 cc., una de ellas con agua destilada y hervida y la otra con la disolución de cloruro potásico. Añadir a cada una 15 g. de suelo desecado al aire y tamizado sobre tamiz de 2 mm. Después de media hora de reposo ce agita suavemente con una varilla, repitiendo esta operación en sentido vertical y circular, dos o tres veces, con intervalos de media hora.
Se deja depositar la tierra durante veinticuatro horas y se lee el volumen de sedimentación.
En los suelos de estructura estable el volumen de sedimentación es el mismo y difieren tanto más cuanto mayor es la inestabilidad.
12.5. Referencia
1. Bouyoucos, G.: «Journ. Am. Soc. Agron.», p. 27, 1935.
Aguas
1. BIOXIDO DE CARBONO LIBRE (CO2)
1.1. Principio
El presente método está basado en la reacción del CO2 libre del agua con el hidróxido de sodio, para formar bicarbonato de sodio.
Además de este método de valoración volumétrica existe uno monográfico que por exigir gran precisión en la determinación «in situ» del pH y de la alcalinidad resulta menos aconsejable.
Pueden producirse errores por la presencia de amoníaco, aminas, fosfatos, boratos, silicatos, sulfuros y nitritos, así como por la existencia de ácidos minerales o sales de ácido fuerte y base débil. También alteran cuantitativamente el resultado el aluminio, hierro, cromo y cobre.
1.2. Material y aparatos
1.2.1. Frascos pyrex de 1.000 ml.
1.2.2. Probeta graduada de 100 ml.
1.2.3. Tubo de goma.
1.2.4. Bureta normal graduada en 0,1 ml.
1.2.5. Varilla de vidrio.
1.3. Reactivos
1.3.1. Solución valorada de hidróxido sódico 0,02 N. Diluir 20 ml. de NaOH 1 N a un litro de agua destilada que previamente se ha hervido no menos de quince minutos, para expulsar el bióxido de carbono y enfriarlo a la temperatura ambiente. Preparar diariamente y proteger del bióxido de carbono atmosférico en un frasco pyrex. Titular la solución con el reactivo 13.3.
1.3.2. Solución indicadora de fenolftaleína. Disolver 0,5 g. de fenolftaleína en 50 ml. de alcohol etílico de 96° y agregar 50 ml. de agua destilada que previamente se ha hervido no menos de quince minutos, para expulsar el bióxido de carbono y enfriarlo a la temperatura ambiente. Agregar, a continuación, el reactivo 1.3.1 a gotas hasta que aparezca una muy ligera coloración rosa.
1.3.3. Solución valorada de bicarbonato sódico 0,01 N. Pesar exactamente 0,84 g. de NaHCO3 anhidro y diluir en 1.000 ml. de agua destilada que previamente se ha hervido no menos de quince minutos, para expulsar el bióxido de carbono y enfriado a la temperatura ambiente. Preparar inmediatamente antes de usarlo.
1.4. Procedimiento
Sifonar la muestra, mediante tubo de goma, llenando desde el fondo una probeta de 100 ml. hasta que derrame. Extraer el tubo de goma y verter el exceso de agua mediante sacudidas.
Agregar cinco o diez gotas del reactivo 1.3.2. Si la muestra vira a rojo, no hay presente bióxido de carbono libre; si la muestra se mantiene incolora, titular rápidamente con el reactivo 1.3.1 agitando suavemente con una varilla de vidrio hasta que el color rosa característico observado a través de todo el espesor de la muestra persista por lo menos treinta segundos.
1.5. Cálculos
Calcular el bióxido de carbono libre, expresado en miligramos por litro, mediante la fórmula

siendo
A = ml. gastados del reactivo 1.3.1.
F = factor de corrección del reactivo 1.3.1 hallado mediante su titulación con el reactivo 1.3.3.
B = ml. de muestra.
1.6. Referencias
1. «Métodos de análisis recomendados». Centro de Estudios Hidrográficos. Madrid, 1968.
2. «Standard Methods for the Examination of Water and Wastewater». American Public Health Association, Inc. New York, 1960.
2. DEMANDA BIOQUIMICA DE OXIGENO (D. B. O.)
2.1. Principio
La demanda bioquímica de oxígeno o D. B. O. es la cantidad de oxígeno eventualmente consumida por los gérmenes aerobios para asegurar la descomposición, en condiciones normalizadas de incubación, de las materias orgánicas contenidas en el agua analizada.
La incubación se efectúa en oscuridad, durante cinco días y a una temperatura de 20° C.
2.2. Material y aparatos
2.2.1. Frascos de incubación, de 250-300 ml. de capacidad, aforados y con tapón esmerilado macizo y con bisel.
2.2.2. Estufa de cultivos con control termostático a 20° C ± 1° C.
2.2.3. Matraces erlenmeyer de 500 ml.
2.2.4. Pipetas de 1 y 5 ml.
2.2.5. Buretas.
2.3. Reactivos
2.3.1. Agua destilada.
2.3.2. Solución amortiguadora de fosfato. Disolver 8,5 g. de KH2PO4, 25,75 g. de K2HPO4, 33,4 g. de Na2HPO4.7H2O y 1,7 g. de NH4Cl en unos 500 ml. de agua destilada y diluir a un litro. El pH de esta solución amortiguadora debe ser de 7,2, sin ajuste alguno.
2.3.3. Solución de sulfato de magnesio. Disolver 22,5 g. de MgSO4. 7H2O en agua destilada y diluir a un litro.
2.3.4. Solución de cloruro de calcio. Disolver 27,5 g. de CaCl2 anhidro en agua destilada y diluir a un litro.
2.3.5. Solución de cloruro férrico. Disolver 0,25 g. de FeCl3. 6H2O en agua destilada y diluir a un litro.
2.3.6. Soluciones de ácidos y álcalis 1 N.
2.3.7. Solución de sulfito de sodio 0,025 N. Disolver 1,575 gramos de NaSO3 anhidro en 1.000 ml. de agua destilada. Esta solución no es estable y se debe preparar el día que se vaya a emplear.
2.3.8. Solución de sulfato manganoso. Disolver 400 g. de MnSO4.2H2O en agua destilada, filtrar y diluir a un litro. Esta solución no debe liberar más que trazas de yodo cuando se añade a una solución acidificada de ioduro potásico.
2.3.9. Reactivo de álcali-ioduro nitruro. Disolver 500 g. de NaOH y 135 g. de NaI en agua destilada. Disolver 10 g. de NaN3 en 40 ml. de agua destilada. Mezclar ambas soluciones y diluir a un litro. Este reactivo no debe producir coloración con el almidón cuando se diluye y acidifica.
2.3.10. Acido clorhídrico concentrado de densidad 22° Beaumé = 1,18.
2.3.11. Solución de almidón. Triturar 5 g. de almidón soluble y 5 mg. de I2Hg con un poco de agua y añadir lentamente la emulsión a un litro de agua hirviendo, manteniendo la ebullición unos minutos hasta que la solución quede clara. Enfriar y pasar a un frasco topacio con tapón esmerilado.
2.3.12. Solución de tiosulfato sódico 0,10 N. Disolver 24,82 g. de Na2S2O3. 5H2O en agua destilada recién hervida y enfriada y diluir a un litro: Conservar la solución añadiendo 1 g. de NaOH.
2.3.13. Solución valorada de dicromato potásico 0,025 N. Pesar 1,226 g. de K2Cr2O7, previamente secados durante dos horas a 103° C y diluir a un litro en matraz aforado.
2.3.14. Solución valorada de tiosulfato sódico 0,025 N. Diluir 250 ml. del reactivo 2.3.12 a un litro de agua destilada recientemente hervida y enfriada. Titular esta solución mediante el reactivo 2.3.13.
2.4. Procedimiento
2.4.1. Preparación del agua de dilución:
— Tomar dos litros de agua destilada.
— Oxigenarla mediante aire comprimido hasta que la tensión del oxigeno disuelto esté próxima a su saturación.
— Añadir a dicha agua 1 ml. de cada uno de los reactivos 2.3.2, 2.3.3, 2.3.4 y 2.3.5.
— Conservar este agua a 20° C tapado el frasco con tapón de algodón.
2.4.2. Acondicionamiento del agua problema:
— Neutralizar el agua problema hasta pH próximo a 7 mediante el reactivo 2.3.6.
— Eliminar el cloro libre, si lo hubiese, mediante la adición del reactivo 2.3.7.
— Eliminar el exceso de oxigeno agitando fuertemente la muestra o bien mediante burbujeo de aire comprimido, siempre a 20° C.
2.4.3. Dilución del agua problema:
— Sifonar el agua de dilución a una probeta graduada de 1.000-2.000 ml. de capacidad, llenándola hasta la mitad sin arrastrar aire. Agregar el agua de muestra cuidadosamente mezclada en la cantidad necesaria para obtener la dilución que se desee y terminar de diluir hasta el nivel apropiado con agua de dilución. Mezclar bien con un agitador, evitando el arrastre de aire.
— Se aconseja las siguientes diluciones:
Efluentes depurados: de 5 a 25 por 100.
Aguas fluviales: de 25 a 100 por 100.
— Sifonar la dilución mezclada a dos frascos de incubación, llenándolos completamente y cerrándolos herméticamente.
— Determinar el oxigeno inicial en uno de los dos frascos de forma inmediata y conservar el otro frasco en incubación durante cinco días en oscuridad y a una temperatura de 20° C para después determinar en él el oxígeno disuelto no consumido.
2.4.4. Determinación del oxígeno disuelto:
— Agregar al frasco de incubación 1 ml. del reactivo 2.3.8 y después 1 ml del reactivo 2.3.9, haciendo ambas adiciones en el fondo del frasco. Tapar de nuevo el frasco, procurando excluir las burbujas, con lo que se derramará una pequeña cantidad del líquido.
— Mezclar por inversión varias veces y esperar que se sedimente el precipitado.
— Destapar el frasco y añadir, mediante pipeta, haciendo llegar el ácido hasta el fondo, 5 ml. de 2.3.10. Cerrar de nuevo el frasco y agitar hasta que se disuelva el precipitado.
— Pasar todo el líquido a un matraz erlenmeyer de 500 ml., lavando frasco y tapón con agua destilada.
— Añadir gota a gota, con rapidez y agitando el matraz, el reactivo 2.3.14 hasta que el líquido tome coloración paja pálido.
— Agregar 1 ó 2 ml. del reactivo 2.3.11, con lo que tomará el liquido color azul, y continuar valorando con 2.3.14, gota a gota, hasta que el líquido se torne incoloro o gris blanquecino.
2.5. Cálculos
Calcular el oxígeno disuelto, expresado en p. p. m., mediante la fórmula

V = ml. de tiosulfato gastados.
f = factor de corrección de la normalidad del tiosulfato hallado mediante la titulación con reactivo 2.3.13.
A = volumen, en ml., del frasco utilizado.
Calcular la D.B.O., expresada en p. p. m., mediante la fórmula

D1 = p. p. m, de O2 disuelto en el frasco no sometido a incubación.
D2 = p. p. m. de O2 disuelto en el frasco sometido a incubación.
C = tanto por ciento del agua problema en el agua problema diluida.
2.6. Observaciones
Este método no es aplicable a aguas negras o desechos industriales concentrados que exigen diluciones muy elevadas y obligan a sembrar el agua de dilución con un inóculo.
En estos casos se precisan comprobaciones de la D.B.O. del inóculo y los métodos y cálculos requieren técnicas laboriosas sólo propias de los laboratorios especializados.
2.7. Referencias
1. «Standard Methods for the examination of water and sewage». American Public Health Association. New York, 1960.
2. Rodier, J.: «L'Analyse chimique et phisico-chimique de l’eau». Dunod, Paris.
3. Gutiérrez Calderón, E., et alt.: «Estudio del B.O.D. y plan de trabajo para su determinación». Anales I.F.I.E. Madrid, 1959.
4. «Livre de l’eau». Cebedoc. Lieja, 1966.
3. CONDUCTIVIDAD ELECTRICA
3.1. Principio
Se denomina conductividad específica de un agua a la aptitud de ésta para transmitir la corriente eléctrica.
La conductividad depende de la actividad y tipo de iones disueltos y de la temperatura a la que se realiza la medida.
Para medir la conductividad se hace uso de un puente de Wheatstone y una célula de conductividad apropiada, comparando a la misma temperatura la resistencia eléctrica de la muestra y de una solución valorada de cloruro potásico y refiriendo el resultado a 25° C.
3.2. Material y aparatos
3.2.1. Puente de Wheatstone apto para este fin.
3.2.2. Celda de conductividad especifica, bien del tipo pipeta, bien del tipo inmersión, con electrodos de platino platinados. La constante de la celda debe ser, aproximadamente, de 1,0 cm‒1 para aguas de baja o moderada conductividad y de 10 cm‒1 para aguas de conductividad alta.
3.3. Reactivos
Solución patrón de ClK. Preparar una solución de CIK cuya conductividad difiera por un factor de menos de 5 de la conductividad de la muestra. Generalmente una solución 0,01 M de CIK (0,7456 g/l es aceptable. Esta solución tiene una conductividad de 1.413 μmho/cm. De la siguiente tabla pueden elegirse otras soluciones patrones que se consideren necesarias:
Conductividad eléctrica de soluciones CIK a 25° C
| Concentración M | Conductividad eléctrica μmho/cm. |
|---|---|
| 10‒4 | 14,94 |
| 5×10‒4 | 73,90 |
| 10‒3 | 147 |
| 5×10‒3 | 717 |
| 10‒2 | 1.413 |
| 2×10‒2 | 2.767 |
| 5×10‒2 | 6.668 |
| 10‒1 | 12.900 |
| 2×10‒1 | 24.820 |
3.4. Procedimiento
Dejar que la solución patrón de CIK y las muestras de agua estén a la misma temperatura. Cualquier temperatura entre 20 y 30° C es aceptable, pero es importante que las soluciones problema y la patrón estén a la misma temperatura. Cuando se requiere gran precisión se colocarán las soluciones a 25° C en termostato.
A continuación se enjuaga y se llena sucesivamente la celda de conductividad con la solución patrón y las soluciones problema y leyendo en el aparato (2-1), de acuerdo con su manual de instrucciones, los valores obtenidos.
3.5. Cálculos

CE (x106) = Conductividad eléctrica.
CEClK= CE (x106) de la solución patrón de CIK empleada.
R = Resistencia en ohmios de la celda con la solución patrón.
R' = Resistencia en ohmios de la celda con la solución problema.
f = Factor de corrección de temperatura de la tabla 3.I.
3.6. Observaciones
3.6.1. Las células nuevas deben limpiarse con mezcla sulfocrómica, debiendo platinarse los electrodos de las mismas cuando los resultados sean erráticos o cuando se observe que el platino se ha desprendido total o parcialmente. Para ello se prepara una solución de 1 g. de cloruro de platino y 0,012 gramos de acetato de plomo en 100 ml. de agua. Se llena la celda con está solución y se conectan los terminales de los electrodos a una batería de 1,5 voltios intercalando un shunt en la batería cuando sea necesario ajustar la corriente, de manera que se desprenda una pequeña cantidad de gas. Dar por terminado el proceso cuando los electrodos estén recubiertos con un depósito de negro de platino. Drenar la célula y recuperar para un futuro uso la solución platonizante Enjuagar bien la celda con agua destilada y dejarla llena de agua destilada cuando no se use.
3.6.2. Hasta 5.000 microohmios existe una aceptable correlación entre conductividad eléctrica y contenido salino de las aguas, de tal modo que los siguientes factores de conversión se manejan frecuentemente.


3.7. Referencias
1. Bower, C. A., y Wilcox, L. V.: «Methods of Soil Analysis Part: 2», pp. 937-940. 1965.
2. National Research Council: «Inter. Critica] tables». 1929.
3. «Agricultural Handboock» núm. 60. USDA. pp. 137-140. 1954.
4. Métodos de análisis recomendados por el Instituto de Hidrología del C.S.I.C., pp. 30-34, 1968.
TABLA 3.1.
Factores de temperatura para corregir los datos de resistencia y conductividad de soluciones acuosas a la temperatura de 25° C
CE25 = CEt × ft : R25 = Rt/ft
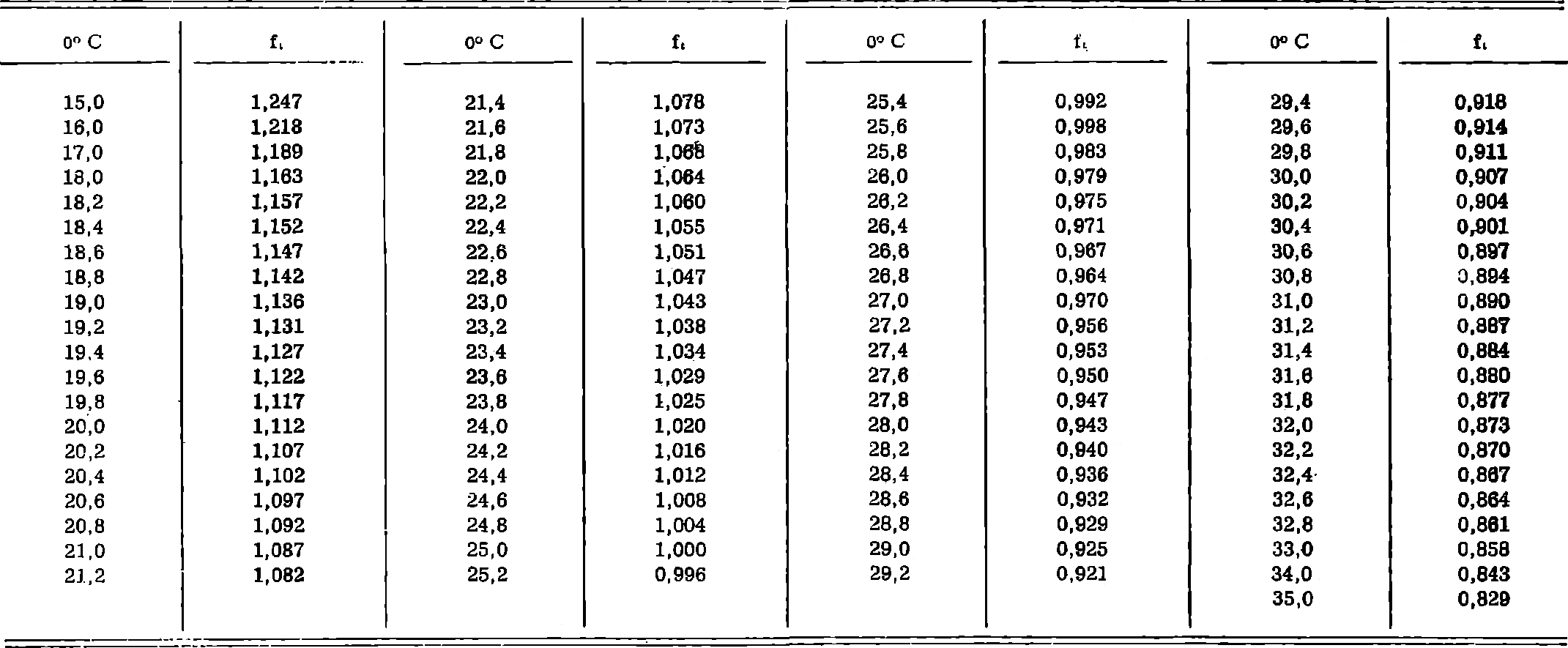
4. SULFATOS
(Determinación turbidométrica)
4.1. Principio
El ion SO4= se precipita con ion Ba2+, en condiciones tales que te formen cristales de tamaño uniformes de SO4Ba los que deben mantenerse en suspensión homogénea durante un período de tiempo que resulte suficiente para medir la absorbancia que la misma produzca.
El contenido en SO4= de cada muestra se obtiene a partir de la curva de calibrado previamente obtenida.
4.2 Material y aparatos
4.2.1. Instrumento de medida, que puede ser uno de los siguientes:
4.2.1.1. Nefeiómetro o turbidimetro.
4.2.1.2. Espectrofotómetro con medida de 420-430 nm.
4.2.1.3. Fotómetro de filtro, equipado con filtro de transmitancia 420-430 nm.
4.2.2. Tubos cilíndricos, calibrados a 50 ml., con tapón.
4.3. Reactivos
4.3.1. Solución de SO4K2 conteniendo 480 mg. (10 meq) de SO4= por litro (0,8707 gramos de SO4K2 disueltos en agua destilada hasta completar un litro).
4.3.2. Solución precipitante de bario. Se disuelven 20 gramos de acetato bárico en una mezcla de 75 ml. de ácido acético 10 N y 25 ml. de solución de goma arábiga al 5 por 100, filtrando la solución resultante. El pH amortiguado de esta solución es de 3,70.
4.4. Procedimiento
4.4.1. Obtención de la curva del calibrado. En los tubos cilíndricos 4.2.2 se introducen partes alícuotas de la solución 4.3.1. Se les añade agua destilada hasta unos 40 mililitro, 1 ml. de reactivo 4.3.2 y se completa a 50 ml. con agua destilada. Se homogeniza el conjunto durante un minuto por agitación suave y se deja en reposo al menos otro minuto. A partir de ese momento, y dentro de los quince minutos siguientes, pueden efectuarse las medidas; para ello, se traslada parte de la suspensión a las cubetas del aparato de medida que se utilice, midiendo las correspondientes absorbancias, empleando como blanco agua destilada sometida al mismo tratamiento.
Con los valores obtenidos se construye la curva de calibrado, que se aconseja tabular.
4.4.2. Valoración de las muestras. Se opera exactamente igual a como se ha indicado en 4.4.1, empleando 5 ml. del problema.
4.5 Cálculo de los resultados
El contenido en SO4= de las muestras se obtiene llevando las lecturas de absorbancias obtenidas con las mismas a la curva de calibrado 4.1 y teniendo en cuenta que se operó con 5 ml. de muestra.
Los resultados pueden expresarse en meq. o en mg. de SO SO4= por litro.
4.6. Observaciones
4.6.1 Si la lectura de una muestra sobrepasa la máxima concentración que figura en la curva de calibrado o inversamente da un valor muy bajo en dicha curva, debe repetirse el ensayo empleando menos de 5 ml. o más de 5 ml. de problema, respectivamente, lo que deberá tenerse en cuenta al efectuar el cálculo del resultado.
4.6.2. No se indican las concentraciones mínimas y máxima, entre las que este método resulta válido, porque ello depende en gran manera del instrumento de medida que se utilice y del paso de luz de la cubeta empleada.
4.6.3. En esta técnica interfieren fundamentalmente el color y la turbiedad. Esta puede eliminarse por filtración o centrifugación. La interferencia del color puede soslayarse utilizando la muestra coloreada como testigo, al que no se le agrega reactivo 4.3.2 o empleando como instrumento de medida un nefelómetro de doble posición de cubeta, con lo que se elimina la influencia del color.
4.7. Referencias
1. Método utilizado en el Laboratorio de Suelos y Aguas del IRYDA. Inf. Quim. Analítica. V. pp. 1-6. 1951
2. Métodos de análisis recomendados por el Instituto de Hidrología del Consejo Superior de Investigaciones Científicas, pp. 11-13, 1968.
5(a) CLORUROS
(Indicador)
5(a).1. Principio
Los procedimientos clásicos para determinación de cloruros se basan en formación de una sal de plata relativamente insoluble.
El punto de viraje de la valoración de cloruros con nitrato de plata puede ser detectado de diversas maneras, tal como por la aparición de un precipitado rojo de CrO4Ag2 (valoración de Mohr) o midiendo el potencial que se desarrolla en la solución mediante una combinación apropiada de electrodos (Kolthoff y Kuroda, 1951).
5(a).2. Material y aparatos
5(a).2.1. Microbureta de 10 ml.
5(a).3. Reactivos
5(a).3.1. Solución al 5 por 100 de cromato potásico. Disolver 5 g. de cromato potásico en 50 ml. de agua y añadir nitrato de plata 1 N, gota a gota, hasta que se forma un precipitado rojo claro permanente. Filtrar y diluir hasta 100 ml.
5(a).3.2. Nitrato de plata, 0,005 N. Disolver 0,8495 g. de secado en estufa de nitrato de plata en agua y diluir exactamente en un litro. Almacenar la solución en un frasco color topacio para protegerla de la luz.
5(a).4. Procedimiento
Tomar una alícuota de la solución problema y valorar con ácido sulfúrico hasta el punto de viraje con naranja de metilo («Métodos Oficiales de Análisis de Aguas», núm. 6). Se usará una solución patrón del ácido en caso de que interese determinar carbonates y bicarbonatos.
Añadir cuatro gotas de reactivo 5(a).3.1 a la muestra guardada después de la determinación de carbonatos-bicarbonatos. Valorar, agitando al mismo tiempo bajo luz brillante, con el reactivo 5(a).3.2, usando una microbureta de 10 ml. hasta que aparezca el primer color rojo (pardo, rojizo) permanente. Las correcciones del ensayo en blanco varían con el volumen de la muestra en el punto de viraje, y generalmente aumenta, regularmente entre 0,03 a 0,20 ml., según aumenta el volumen de 2 a 12 ml.
5(a).5. Cálculos
Calcular la concentración de cloruros en meq/l.

V = volumen, en ml., de NO3Ag utilizados en la valoración de la solución problema.
V' = volumen, en ml., de NO3Ag utilizados en la valoración en blanco.
V'' = volumen, en ml., de alícuota de solución problema.
5(a).6. Observaciones
Los ácidos minerales que disuelven los cromatos de plata deben de estar ausentes. Por tanto, la cantidad de ácido añadida debe ser la estrictamente necesaria para hacer la solución débilmente ácida, evitándose la adición en exceso. Los ioduros, bromuros y bicarbonatos forman precipitados con nitrato de plata y deben estar ausentes de la muestra. Generalmente, los fosfatos no interfieren pero también deben de evitarse cantidades altas de fosfatos. Los materiales orgánicos también deben de eliminarse por su tendencia a reducir el nitrato de plata en soluciones neutras. El hierro, normalmente, reacciona con el cromato potásico produciendo un cromato insoluble, pero la cantidad de hierro que generalmnete se encuentra en las muestras no interfiere. En caso de que se crea que la cantidad de hierro es alta, añadir unas cuantas gotas más de indicador para asegurar el exceso de cromato.
5(a).7. Referencias
1. Chapman, H. D., y Pratt, P. F.: «Methods of Analysis for Soils, Plants and Water», pp. 98-100, 1961.
2. Reitmeier, R. F.: «Semimicroanalysis of Saline oil Solutions». Indus. and Engin. Chem. Analyt. Ed. 15: pp. 393-402, 1943.
3. Stout, P. R., y Johnson, C. M.: «Methods of Soils Analysis». Part 2: pp. 1.125-1.126. American Society of Agronomy, 1965.
5(b) CLORUROS
(Potenciometría)
5(b).1. Principio
Ver 5(a).1.
5(b).2. Material y aparatos
5(b).2.1. Potenciómetro (pH-metro).
5(b).2.2. Electrodos de vidrio.
5(b).2.3. Electrodo de plata-cloruro de plata (Ag-ClAg).
5(b).2.4. Microbureta de 5 ó 10 ml.
5(b).2.5. Agitador magnético.
5(b).3. Reactivos
5(b).3.1. Suspensión de cloruro de plata (ClAg). Preparar un precipitado de ClAg mezclando soluciones 0,1 N de CINa y NO3Ag; 500 ml. de cada solución serán suficientes para precipitar ClAg para muchas determinaciones. Debe de usarse un ligero exceso de cloruro o de nitrato para provocar un buen precipitado. Lavar bien el precipitado con agua destilada y transferirlo a un frasco topacio. Renovar la solución sobrenadante diariamente durante diez días, reemplazándola con agua destilada para remover las trazas de los excesos de iones planta o cloruro. Finalmente, completar el volumen con agua hasta un litro.
5(b).3.2. Solución de nitrato de plata (NO3Ag) 0,1 N. Disolver 8,5 g. de NO3Ag en agua destilada, diluir la solución en un volumen de 500 ml. y almacenar en un frasco topacio.
5(b).3.3. Solución de nitrato de plata (NO3Ag) 0,01 N. Diluir 10 ml. de reactivo 5(b).3.2 en 100 ml. con agua destilada. Comprobar antes de usarla su normalidad valorándola potenciométricamente con la solución ClK [reactivo 5(b).3.4], según se describe más adelante.
5(b).3.4. Solución patrón de cloruro potásico (ClK) 0,01 N. Disolver 0,746 g. de ClK puro y desecado en estufa y diluir hasta un litro. Esta solución es 0,01 N con respecto al cloruro y se usa para valorar la solución de NO3Ag.
5(b).3.5. Solución soporte de electrolito. Disolver 101g. de nitrato potásico (NO3K) recristalizado (calidad reactivo) en agua, añadir 62 ml. de ácido nítrico concentrado (NO3H) y diluir la solución en un litro.
5(b).4. Procedimiento
Transferir una alícuota de la solución problema preferentemente que contenga menos de 0,1 meq. de cloruros al vaso de valoración. Diluir en agua destilada hasta un total de 25 ml., aproximadamente. Añadir 2,5 ml. de la solución de electrolito.
Preparar la solución de referencia añadiendo a 25 ml. de agua destilada, 2,5 ml. de la solución soporte de electrolito y dos gotas de la suspensión de ClAg. Sumergir el juego de electrodos de referencia, agitando ligeramente, cerrar el circuito del pH-metro y anotar el potencial observado en milivoltios. Reemplazar la solución de referencia por la solución problema que contenga la solución soporte de electrolito. Valorar agitando suavemente con NO3Ag 0,01 N hasta que el potencial que indique la escala del potenciómetro o pH-metro sea el mismo que el anotado para la solución de referencia. Anotar el volumen de solución de NO3Ag requerido.
5(b).5. Cálculo
Calcular la concentración de Cl‒ en la solución problema, expresada en meq/I.

V = volumen, en ml., de la solución de NO3Ag.
N = normalidad de la solución de NO3Ag.
V' = volumen, en ml., de la solución problema.
5(b).6. Observaciones
5(b).6.1. Los vasos de valoración no deben exponerse a la luz solar directa durante la valoración, lo que podría provocar un cambio de potencial final. La agitación excesivamente rápida también puede cambiar el potencial final añadiendo una componente de potencial de corriente.
5(b).6.2. El uso de la solución soporte de electrolito de alta fuerza iónica dispensa de conocer al verdadero potencial de equivalencia al mismo tiempo que ahorra las correcciones necesarias por diferencia de fuerza iónica de la solución problema o clase de electrolitos disueltos. El «potencial aparente de equivalencia» que se obtiene es prácticamente independiente de las variaciones normales al diferente contenido de sales solubles de la solución problema.
5(b).6.3. El procedimiento descrito no diferencia entre los iones cloruros y bromuros.
5(b).6.4. Existen en el mercado diversos equipos de valoración de cloruros en los que la valoración queda automáticamente interrumpida cuando se produce un cambio brusco de potencial o conductividad que equivale a la precipitación de todos los iones Cl‒ inicialmente presentes como ClAg. Simultáneamente se expresa digitalmente la concentración de cloruros en la alícuota añadida a la solución soporte, en la que se realiza la valoración.
5(b).7. Referencias
1. Fisher, R. B., y Peter, D. G.: «Quantitatives Chemical analysis». Saunders, 1968.
2. Golterman, H. L.: «Methods for Chemical analysis of fresh waters». TBP Hb60. Blacwell Oxford. 1970.
3. Kolthoff, I. M., y Kurola, P. K.: «Determination of traces of chloride». Anal. Chem. 23: pp. 1.304-1.306, 1951.
4. Stout, P. R., y Johnson, C. M.: «Methods of Soils Analysis». Part 2, pp. 1.127-1.129. American Society of Agronomy, 1965.
6. ALCALINIDAD
6.1. Principio
Se determina por valoración con una solución valorada en en un ácido mineral fuerte a los puntos de equivalencia del bicarbonato (pH 8,3) y ácido carbónico (entre pH 4,2 y 5,4), bien sea potenciométricamente o por medio de indicadores. El segundo punto de equivalencia indica la alcalinidad total de la muestra que puede ser debida a los siguientes iones: hidróxido, carbonatos y bicarbonatos. El pH al que se produce dicha equivalencia depende de la cantidad de CO2 en solución. Para una muestra dada, la eliminación del CO2 en solución cuando la valoración se realice en atmósfera de nitrógeno o cuando se hierve en las proximidades del punto de viraje para suprimir el CO2 produce un cambio más brusco del potencial o del color, pero la posición del punto de viraje no cambia.
Suponiendo que las cantidades presentes en el agua de Si O4H3, BO3H2, NH4, SH‒ y CO3Ca coloidal o en suspensión sean despreciables, con las valoraciones mencionadas se pueden determinar las concentraciones individuales de hidróxidos, carbonatos y bicarbonatos.
6.2. Material y aparatos
6.2.1. Matraces erlenmeyer de 200 ml.
6.2.2. Pipeta.
6.2.3. Bureta.
6.2.4. Potenciómetro.
6.3. Reactivos
6.3.1. Acido sulfúrico o clorhídrico 0,1 N o 0,05 N, según la alcalinidad prevista de la muestra.
6.3.2. Indicador de fenolftaleína: Disolver 0,5 g de fenolftaleína en 50 ml. de etanol de 95 por 100 y añadir 50 ml. de agua. Añadir gota a gota solución de NaOH 0,05 N libre de CO2 hasta que el color se vuelva débilmente rosado.
6.3.3. Indicador de anaranjado de metilo al 0,05 por 100 de agua.
6.4. Procedimiento
Tomar una alícuota que contenga menos de 1 meq. de alcalinidad total y diluir hasta 50 ml. con agua si fuese necesario. Añadir dos gotas de 6.3.2 si se produce color, valorar con 6.3.1 hasta que desaparezca el mismo. Añadir dos gotas de 6.3.3 y continuar la valoración con 6.3.2 hasta el punto de viraje del anaranjado de metilo. Caso de que exista dificultad en apreciar el viraje, detener la valoración en las proximidades del mismo, hervir el matraz durante unos minutos, dejar enfriar y continuar la valoración.
Hacer un ensayo en blanco con los reactivos y 50 ml. de agua y restar de los anteriores valores los encontrados en este ensayo, caso de que fuesen apreciables.
En caso de utilizar potenciómetro, la primera valoración de la muestra se hará hasta pH = 3,8 y la segunda hasta pH = 4,0.
6.5. Cálculos
a = ml. de alícuota de la solución problema.
x = ml. la lectura de la bureta en el punto de viraje de la fenolftaleína después de corregir por ensayo en blanco.
z = ml. la lectura de la bureta en el punto de viraje del anaranjado de metilo después de corregir por ensayo en blanco.
N = normalidad del ácido.
Caso 1.º x < ½ z



Caso 2.º x > ½ z



Caso 3.º x = ½ z
Concentraciones meq/l.
(OH‒) y (CO3H‒) despreciables, aplicar para (CO3=) cualquiera de las dos igualdades del caso 1.º y 2.º
Todos los casos

6.6. Observaciones
Todos los reactivos, así como el agua usada en diluciones y ensayos en blanco, deben obtener un bajo contenido en CO2, particularmente cuando se trate de muestras con alcalinidad baja. La eliminación del CO2 disuelto en el agua puede conseguirse mediante cualquiera de los dos procedimientos siguientes:
1. Reducir la presión durante diez a quince minutos con una trompa de agua.
2. Hervir durante diez a quince minutos y dejar enfriar en un erlenmeyer con tapón.
6.7. Referencia
1. Golterman, H. L.: «Methods for Chemical analysis of fresh waters». IBP Handbook no. 8 Black Well Scientif Publications. Oxford, 1970.
Agence d'État Bulletin Officiel de l'État
Av. Manoteras, 54 - 28050 Madrid




