Agencia Estatal Boletín Oficial del Estado






La disposición final primera del Real Decreto 1329/1995, de 28 de julio, por el que se fijan líneas directrices para la evaluación de los aditivos en la alimentación animal, faculta al Ministro de Agricultura, Pesca y Alimentación, previo informe favorable del Ministerio de Sanidad y Consumo, para dictar, en el ámbito de sus competencias, las disposiciones necesarias para su aplicación y, en especial, para adaptar el contenido de su anexo a las correspondientes modificaciones de la normativa comunitaria, en función de los avances de los conocimientos científicos y técnicos.
De acuerdo con ello, la presente Orden incorpora al ordenamiento jurídico la Directiva 2001/79/CE, de la Comisión, de 17 de septiembre, por la que se modifica la Directiva 87/153/CEE, del Consejo, por la que se fijan líneas directrices para la evaluación de los aditivos en la alimentación animal.
La resistencia bacteriana a los antibióticos, causada por su utilización como aditivos en la alimentación animal, va en aumento y constituye un problema fundamental para la salud pública. Por ello, es preciso complementar las directrices para aditivos distintos de los microorganismos y las enzimas, con objeto de incluir en el expediente de autorización de aditivos para la alimentación animal una evaluación del riesgo de selección o transferencia de la resistencia a los antibióticos y de cualquier aumento de la persistencia y excreción de patógenos intestinales, con el fin de garantizar la inocuidad de dichos aditivos.
Asimismo, se evidencia la necesidad de evaluar las repercusiones medioambientales de los aditivos de los piensos incluyendo, en las directrices mencionadas, criterios de evaluación del riesgo medioambiental. Del mismo modo, en las directrices se añade mayor información sobre la manera en que los trabajadores y los utilizadores pueden verse expuestos a los aditivos.
En la elaboración de la presente Orden han sido consultados los sectores afectados y las Comunidades Autónomas.
Esta disposición ha sido informada favorablemente por la Comisión Interministerial para la Ordenación Alimentaria.
En su virtud, previo informe favorable del Ministerio de Sanidad y Consumo, dispongo:
Se sustituye el anexo del Real Decreto 1329/1995, de 28 de julio, por el que se fijan líneas directrices para la evaluación de los aditivos en la alimentación animal por el anexo de la presente disposición.
La presente Orden entrará en vigor el día siguiente al de su publicación en el «Boletín Oficial del Estado».
Madrid, 31 de enero de 2002.
ARIAS CAÑETE
Parte I: Aditivos distintos de los microorganismos y las enzimas
Consideraciones generales
Las presentes líneas directrices constituyen una guía para la confección de los expedientes de las sustancias y preparados en relación a los que se solicita autorización para su uso como aditivos en la alimentación del animal, o para solicitar autorización para un nuevo empleo de un aditivo ya autorizado. En estas líneas directrices, el término «aditivo» se refiere a las sustancias químicamente activas especificadas o a los preparados que contengan sustancias activas, en la forma en que esas sustancias son incorporadas a las premezclas y a los piensos. Los expedientes deberán permitir evaluar los aditivos de conformidad con los conocimientos actuales y comprobar que responden a los principios fundamentales que requiere para su autorización el artículo 4 del Real Decreto 2599/1998, de 4 de diciembre, sobre los aditivos en la alimentación de los animales.
Los expedientes relativos a aditivos que contengan o estén constituidos por organismos modificados genéticamente según lo dispuesto por la Ley 15/1994, de 3 de junio, por la que se establece el régimen jurídico, utilización confinada, liberación voluntaria y comercialización de organismos genéticamente modificados, contendrán, además de la información requerida por esta norma, la información adicional especificada en el apartado 1 del artículo 6 del Real Decreto 2599/1998, de 4 de diciembre, sobre los aditivos en la alimentación de los animales.
Los expedientes contendrán informes detallados de todos los estudios realizados, presentados según el orden y la numeración propuestos en estas directrices. Asimismo, contendrán referencias y copias de todos los datos científicos publicados pertinentes para la evaluación del aditivo. Deberá haber una versión electrónica del expediente disponible. Los estudios estarán dirigidos a demostrar que el uso del aditivo es seguro en relación con:
a) Las especies diana, en los niveles propuestos para la incorporación en los piensos;
b) Para las personas que, al manejar el aditivo, ya sea en forma pura o incorporado a las premezclas o los piensos, estén expuestas al aditivo a través de las vías respiratorias o por contacto con otras glándulas mucosas, los ojos o la piel;
c) Para los consumidores que ingieren alimentos procedentes de animales a los que se ha administrado el aditivo y que podrían contener restos del aditivo o de sus metabolitos; en general, esto se asegurará por medio del establecimiento de plazos de retirada y límites máximos de residuos (LMR);
d) Para los animales y las personas a través de la selección y la difusión de genes resistentes a los agentes antimicrobianos;
e) En relación con los efectos sobre el medio ambiente del propio aditivo o de los productos derivados del mismo, ya sea directamente o a través de los excrementos de los animales.
Como norma general, deberán presentarse los estudios tendentes a establecer la identidad, las condiciones de empleo, las propiedades fisicoquímicas, los métodos de control y la eficacia del aditivo así como los efectos biológicos y toxicológicos de su evolución metabólica y sus residuos en las especies diana. Cuando el aditivo se destine a una categoría de animales pertenecientes a una especie determinada, deberán realizarse estudios relativos a la eficacia y a los residuos con esa misma categoría. Los estudios necesarios para evaluar los riesgos para la salud humana y el medio ambiente dependerán fundamentalmente del tipo de aditivo y de las circunstancias de su empleo. A este respecto, no se aplicará ninguna norma estricta. En caso necesario, se requerirá información adicional. Deberá justificarse la omisión en el expediente de cualquiera de los datos previstos en las presentes líneas directrices. En particular, sólo se podrá prescindir de estudios sobre el carácter mutagénico y carcinogénico y la toxicidad reproductiva si de la composición química, la experiencia práctica u otras consideraciones se deriva que es razonable excluir estos efectos.
Los estudios se realizarán y presentarán de conformidad con las normas de calidad adecuadas (por ejemplo, las prácticas correctas de laboratorio descritas en el Real Decreto 2043/1994, de 14 de octubre, relativo a la inspección y verificación de buenas prácticas de laboratorio).
Se presentarán informes de expertos en relación con la calidad, la eficacia y la seguridad. Sus autores, que estarán debidamente cualificados y serán expertos reconocidos en el ámbito en cuestión, no podrán participar personalmente en la realización de las pruebas incluidas en el expediente. Los informes incluirán una evaluación crítica de la documentación suministrada por el solicitante; un resumen objetivo no será suficiente.
La determinación de las propiedades fisicoquímicas, toxicológicas y ecotoxicológicas se llevará a cabo utilizando los métodos fijados en el Real Decreto 363/1995, de 10 de marzo, por el que se aprueba el Reglamento sobre notificación de sustancias nuevas y clasificación, envasado y etiquetado de sustancias peligrosas, o por medio de métodos internacionalmente reconocidos por organismos científicos. El uso de métodos distintos de los mencionados deberá justificarse debidamente.
Los expedientes contendrán un resumen apropiado, una propuesta de anexo y, de forma optativa, una monografía. Los expedientes relativos a los antibióticos, los coccidiostáticos y otras sustancias medicamentosas, y a los factores de crecimiento, se acompañarán de una monografía conforme al modelo de la sección V, que permita identificar y caracterizar el aditivo en cuestión, de conformidad con el artículo 3 del Real Decreto 2599/1998, de 4 de diciembre, sobre los aditivos en la alimentación de los animales. Para todos los aditivos se suministrará una ficha descriptiva conforme al modelo de la sección VI.
No siempre será necesario someter los aditivos destinados exclusivamente a la alimentación de animales de compañía a pruebas de toxicidad crónica, mutagénesis, toxicidad reproductiva carcinogénesis tan exhaustivas como las requeridas para los aditivos destinados a la alimentación de animales de abasto, de los que se obtienen productos destinados al consumo humano. No se requieren estudios relativos a los residuos en relación con los animales de compañía.
El estudio de la evolución metabólica del aditivo en los animales diana destinados a la producción de alimentos y en las especies de laboratorio utilizadas para las pruebas de toxicidad se considera necesario a fin de:
a) Asegurar que haya una información adecuada en relación con la toxicidad del aditivo principal y de cualquier metabolito que se forme en las especies a las que pueda estar expuesto el consumidor. A este fin, es importante llevar a cabo una comparación de las transformaciones metabólicas del aditivo en las especies animales utilizadas para el consumo humano y en las pruebas de toxicidad realizadas en el laboratorio;
b) Identificar y cuantificar los residuos marcadores adecuados, así como los plazos de retirada del producto final.
Índice
1. Capítulo I: Resumen de los datos del expediente.
2. Capítulo II: Identidad, características y condiciones de empleo del aditivo; métodos de control.
2.1 Identidad del aditivo.
2.2 Caracterización de la(s) sustancia(s) activa(s).
2.3 Caracterización del aditivo: propiedades fisicoquímicas y tecnológicas.
2.4 Condiciones de empleo del aditivo.
2.5 Métodos de control.
3. Capítulo III: Estudios sobre la eficacia del aditivo.
3.1 Estudios relativos a los efectos en los piensos.
3.2 Estudios de los efectos en los animales.
3.3 Estudios de la calidad de los productos animales.
3.4 Estudios de los efectos sobre las características de los residuos de origen animal.
4. Capítulo IV: Estudios sobre la seguridad de empleo del aditivo.
4.1 Estudios sobre las especies diana.
4.2 Estudios con animales de laboratorio.
4.3 Evaluación de la seguridad para el consumo humano.
4.4 Evaluación de la seguridad de los trabajadores.
4.5 Evaluación del riesgo para el medio ambiente.
5. Capítulo V: Modelo de monografía.
5.1 Identidad del aditivo.
5.2 Características de la sustancia activa.
5.3 Propiedades fisicoquímicas, tecnológicas y biológicas del aditivo.
5.4 Métodos de control.
5.5 Propiedades biológicas del aditivo.
5.6 Indicación cualitativa y cuantitativa de eventuales residuos encontrados en productos de origen animal después de haber seguido las condiciones de empleo del aditivo previstas.
5.7 Si procede, se proporcionarán el valor IDA, los LMR establecidos y el periodo de espera.
5.8 Otras características pertinentes para la identificación del aditivo.
5.9 Condiciones de uso.
5.10 Fecha.
6. Capítulo VI: Modelo de ficha descriptiva.
7. Capítulo VII: Renovación de la autorización de los aditivos cuya autorización está vinculada a un responsable de su puesta en circulación.
8. Capítulo VIII: Nueva solicitud basada en la primera autorización de un aditivo cuya autorización está vinculada a un responsable de su puesta en circulación.
1. Capítulo I: Resumen de los datos del expediente
El resumen seguirá el orden establecido en las directrices y en las diferentes partes hará referencia a las páginas pertinentes del expediente. Contendrá una propuesta que cubra todas las condiciones para la autorización solicitada.
2. Capítulo II: Identidad, características y condiciones de empleo del aditivo; métodos de control
2.1 Identidad del aditivo.
2.1.1 Nombre(s) del propietario propuesto.
2.1.2 Tipo de aditivo según su función principal. Si es posible, se incluirán pruebas del/de los modo(s) de acción. Se especificará cualquier otro uso de la sustancia activa.
2.1.3 Composición cuantitativa y cualitativa (sustancia activa, otros componentes, impurezas, variación de un lote a otro). Si la sustancia activa es una mezcla de componentes activos, deberán describirse por separado los principales, indicándose su proporción en la mezcla.
2.1.4 Estado físico, granulometría, forma de las partículas, densidad, densidad aparente; para líquidos: viscosidad, tensión superficial.
2.1.5 Procedimiento de fabricación. Eventuales tratamientos específicos.
2.2 Caracterización de la sustancia o sustancias activas.
2.2.1 Denominación genérica, denominación química conforme a la nomenclatura de la UIQPA (Unión Internacional de Química Pura y Aplicada), otras denominaciones y abreviaturas genéricas internacionales. Número CAS (Chemical Abstract Service Number).
2.2.2 Fórmula desarrollada, fórmula molecular, peso molecular. Sustancias activas resultado de un proceso de fermentación: origen microbiano (nombre y lugar de la colección de cultivos reconocida como autoridad internacional de depósito donde se encuentre depositada la cepa, si es posible una colección de la Unión Europea, número de registro y todas las propiedades morfológicas, fisiológicas, genéticas y moleculares pertinentes para su identificación). En el caso de cepas modificadas genéticamente, se proporcionará información relativa a la modificación genética en cuestión.
2.2.3 Pureza, identificación y cualificación de las impurezas químicas y microbianas y de las sustancias tóxicas existentes, confirmación de la ausencia de organismos de producción.
2.2.4 Propiedades pertinentes. Propiedades físicas de las sustancias químicamente definidas: constante de disociación, valores de pKa, propiedades electrostáticas, punto de fusión, punto de ebullición, densidad, presión de vapor, solubilidad en agua y en disolventes orgánicos, Kow y Koc, espectro de masa y de absorción, datos relativos al NMR, posibles isómeros y cualquier otra propiedad física pertinente.
2.2.5 Procedimientos de obtención y purificación, medios empleados y, para los productos de fermentación, variación en la composición de los lotes.
2.3 Caracterización del aditivo: propiedades fisicoquímicas y tecnológicas.
2.3.1 Estabilidad de cada fórmula del aditivo respecto a la exposición a agentes atmosféricos como la luz, la temperatura, el pH, la humedad, el oxígeno y el material de envasado. Período de validez para su comercialización.
2.3.2 Estabilidad de cada fórmula del aditivo durante la preparación y el almacenamiento de las premezclas y los piensos, en particular, estabilidad frente a los procesos operacionales y las condiciones de almacenaje previstos (calor, humedad, esfuerzo de cizallamiento, tiempo y material de envasado). Posible degradación o descomposición de los productos. Período de validez del aditivo.
2.3.3 Otras propiedades fisicoquímicas y tecnológicas pertinentes para la obtención y conservación de mezclas homogéneas en las premezclas y en los piensos, propiedades electrostáticas y relativos a la formación de polvo y capacidad de dispersión de los líquidos.
2.3.4 Incompatibilidades o interacciones previstas con piensos, excipientes, otros aditivos aprobados o medicamentos.
2.4 Condiciones de empleo del aditivo.
2.4.1 Los aditivos que tengan efectos tecnológicos y zootécnicos importantes deben cumplir los requisitos de ambas solicitudes. Las solicitudes relativas a cada aditivo deberán identificarse y justificarse.
2.4.2 Empleo tecnológico propuesto en la fabricación de piensos o, en su caso, de materias primas.
2.4.3 Empleos previstos en la alimentación animal (por ejemplo, especies o categorías de animales, grupo de edad o fase de producción del animal, tipo de pienso y contraindicaciones).
2.4.4 Métodos y niveles propuestos de inclusión en premezclas y piensos, o, en su caso, materias primas, expresados como porcentaje del aditivo y sustancias químicamente especificadas en términos de peso en premezclas, piensos o, en su caso, materias primas, incluida la dosis propuesta en el pienso final y la duración propuesta para la administración y, en su caso, el plazo de retirada.
2.4.5 Se proporcionarán datos sobre otros empleos conocidos de la sustancia activa (por ejemplo en alimentos, medicina humana o veterinaria, agricultura e industria).
2.4.6 La ficha de datos de seguridad propuesta de acuerdo con las disposiciones del Real Decreto 1078/1993, de 2 de julio, por el que se aprueba el Reglamento sobre clasificación, envasado y etiquetado de preparados peligrosos, y, en su caso, las medidas propuestas para la prevención de los riesgos laborales y los medios propuestos de protección durante los procesos de fabricación, manipulación, utilización y eliminación.
2.5 Métodos de control.
2.5.1 Descripción de los métodos empleados para establecer los criterios enunciados en los puntos 2.1.3, 2.1.4, 2.2.3, 2.2.4, 2.3.1, 2.3.2, 2.3.3 y 2.3.4.
2.5.2 Descripción de los métodos de análisis cualitativo y cuantitativo utilizados en el control rutinario de la sustancia activa en las premezclas y los piensos. Este método deberá validarse con una prueba del anillo en la que participen por lo menos cuatro laboratorios o se validará internamente de acuerdo con las directrices internacionales armonizadas para la validación interna de métodos de análisis1 en relación con los siguientes parámetros: aplicabilidad, selectividad, calibración, exactitud, precisión, intervalo, límite de detección, límite de cualificación sensitiva, robustez y practicabilidad. Deberán proporcionarse pruebas de que estas características han sido evaluadas (2.5.4).
1 Method Validation-A Laboratory Guide, EURACHEM Secretariat, Laboratory of the Government Chemist, Teddington, UK, 1996.
2.5.3 Descripción de los métodos de análisis cualitativo y cuantitativo utilizados para determinar el residuo o residuos marcadores2 de la sustancia activa en los tejidos o los productos animales en cuestión.
2 El residuo marcador es un residuo cuya concentración se encuentra en una relación conocida con el índice en el que la concentración total de residuos en el tejido en cuestión sobrepasa el LMR.
2.5.4 Los métodos mencionados en los puntos 2.5.2 y 2.5.3 deben ir acompañados de información relativa al método de muestreo aplicado, el porcentaje de recuperación, la especificidad, la exactitud, los límites de detección, los límites de cuantificación y el método de validación utilizado. Los patrones de referencia de la sustancia activa y del/de los residuo(s) marcador(es) deben estar disponibles, así como información relativa a las condiciones de almacenamiento óptimas para estos patrones de referencia. A la hora de concebir los métodos se debe tener en cuenta que los límites de cuantificación deben ser inferiores a los LMR. Además, debe tenerse en cuenta su idoneidad para realizar análisis rutinarios.
3. Capítulo III: Estudios sobre la eficacia del aditivo
3.1 Estudios relativos a los efectos en los piensos. Estos estudios tendrán por objeto los aditivos tecnológicos, tales como los agentes antioxidantes, conservantes, aglutinantes, emulsionantes, estabilizadores, gelificantes, modificadores de pH, etc., que se destinen a mejorar o estabilizar las características de las premezclas y de los piensos pero que no tienen efectos biológicos directos en los productos de origen animal. Todas las actividades o efectos del aditivo que se presenten deberán estar justificados a través de información científica.
Deberá probarse la eficacia del aditivo por medio de los criterios adecuados establecidos en métodos reconocidos aceptables en las condiciones de empleo previstas en comparación con piensos de control adecuados. Estos estudios se elaborarán y se realizarán de modo que permitan una evaluación estadística.
Se proporcionará información completa sobre las sustancias activas, los preparados, las premezclas y los piensos analizados, el número de referencia de los lotes y las condiciones detalladas del tratamiento y las pruebas. Para cada estudio se describirán los efectos positivos y negativos, tanto biológicos como tecnológicos.
3.2 Estudios de los efectos en los animales. Los estudios relativos a los aditivos zootécnicos se realizarán comparando cada una de las especies o las categorías animales a las que se destine el aditivo con grupos de control negativo (sin antibióticos, promotores del crecimiento u otras sustancias medicamentosas) y, eventualmente, con grupos de animales a los que se administran piensos que contengan aditivos aprobados en la UE, de reconocida eficacia en las dosis recomendadas (control positivo).
Los animales utilizados estarán sanos y formarán preferiblemente un grupo homogéneo.
Los estudios permitirán evaluar la eficacia del aditivo de acuerdo con las prácticas ganaderas de la UE. En la medida de lo posible, los protocolos utilizados en todas las pruebas tendrán el mismo diseño, a fin de que, en su caso, se pueda probar la homogeneidad de los datos y se puedan utilizar éstos (si las pruebas así lo indican) para una evaluación estadística.
No se recomienda ningún método en particular, al permitirse la flexibilidad en la elección de criterios científicos al diseñar y realizar los estudios. El diseño experimental utilizado se justificará de acuerdo con el empleo propuesto del aditivo e incluirá consideraciones relativas a su idoneidad estadística.
3.2.1 En relación con los coccidiostáticos y otras sustancias medicamentosas. Deberá atenderse, en primer lugar, a la comprobación de los efectos específicos (por ejemplo, especies controladas, fases afectadas del ciclo vital) y, especialmente, a las propiedades profilácticas (por ejemplo, morbilidad, mortalidad, número de ooquistes y calificación de las lesiones).
Se adjuntará información sobre la eficacia nutritiva y sobre el crecimiento de los animales.
Los datos sobre la eficacia requeridos hacen referencia a tres tipos de investigación de los animales diana:
a) Experimentos controlados en jaulas en batería (infecciones simples y compuestas);
b) Estudios controlados en corral (simulación de las condiciones de empleo);
c) Estudios de campo controlados (condiciones de empleo reales).
Simultáneamente, y si ello fuera pertinente, en las pruebas de eficacia se registrará información adicional a fin de facilitar una evaluación de las interferencias en el crecimiento y en la transformación de los piensos (aves de abasto, gallinas ponedoras y conejos) y los efectos en la fertilidad y fecundidad de los huevos (aves de cría).
3.2.2 En relación con otros aditivos zootécnicos. Se facilitará información sobre los efectos en la alimentación, el crecimiento, la eficacia nutritiva (preferiblemente sobre una base sólida), la calidad y el rendimiento del producto y cualquier otro parámetro que tenga efectos positivos sobre los animales, el medio ambiente, el productor o el consumidor. En su caso, los estudios incluirán una indicación de la relación dosis/respuesta.
3.2.3 Condiciones experimentales. Las pruebas se realizarán por lo menos en dos lugares diferentes. Se describirán por separado y proporcionarán detalles de los controles y de cada tratamiento experimental. El protocolo de las pruebas se elaborará cuidadosamente, teniendo en cuenta los siguientes datos descriptivos generales:
3.2.3.1 Rebaño o piara: Lugar y tamaño; condiciones de alimentación y de crianza, y método de alimentación; para las especies acuáticas, tamaño y número de tanques o cisternas de la explotación y calidad del agua.
3.2.3.2 Animales: especie (las especies acuáticas destinadas al consumo humano se identificarán mediante su nombre corriente, seguido de su denominación latina o Linnean entre paréntesis), raza, edad, sexo, método de identificación, fase fisiológica y estado general de salud.
3.2.3.3 Número de los grupos de experimentación y de control; número de animales en cada grupo: El número de animales que participan en las pruebas permitirá el análisis estadístico. Se establecerán los métodos de evaluación estadística. Para mostrar el efecto mencionado se proporcionarán como mínimo tres (3) pruebas independientes comparables a nivel de p < 0,05 en cada una de las categorías de animales propuestas. En el caso de los rumiantes, se podrá aceptar un nivel de probabilidad más bajo, p < 0,10. El informe incluirá todos los animales o unidades experimentales que ya han participado en las pruebas. Se informará de los casos que no se puedan evaluar debido a la falta o la pérdida de información, así como su distribución en los grupos de animales clasificados.
3.2.3.4 Dietas: Descripción de la fabricación y la composición cuantitativa de la dieta o dietas en términos de ingredientes utilizados, nutrientes pertinentes (valores analizados) y energía. Registros de alimentación.
3.2.3.5 Concentración en los piensos de la sustancia activa (y, en su caso, de las sustancias utilizadas como elemento de comparación), determinada mediante un análisis de control con el método reconocido apropiado: Número de referencia de los lotes.
3.2.3.6 Fecha y duración exacta de las pruebas: Fecha y tipo de los exámenes realizados.
3.2.3.7 Estudios del establecimiento de la dosis: El objetivo de estos estudios es explicar los motivos que han conducido a afirmar que una dosis o una serie de dosis determinada(s) presenta(n) una eficacia óptima. La determinación de la dosis se hará sobre la base de un control (sin antibióticos, promotores del crecimiento u otras sustancias medicamentosas) y como mínimo a tres niveles distintos de cero en los animales diana.
3.2.3.8 Se comunicará la fecha y la ocurrencia de toda consecuencia negativa del tratamiento sobre individuos o grupos (detalles del programa de observación aplicado en el estudio).
3.2.3.9 Todos los aditivos estudiados en condiciones de explotación dispondrán de pruebas científicas válidas en relación con la seguridad para el usuario, el consumidor, el animal y el medio ambiente. Si un aditivo no cumple los requisitos de seguridad de los consumidores, los estudios sobre el mismo se diseñarán para prevenir que los productos animales derivados de los animales de prueba se introduzcan en la cadena de alimentación humana.
3.3 Estudios de la calidad de los productos animales. En su caso, se examinarán las características organolépticas, nutritivas, higiénicas y tecnológicas de los productos de origen animal.
3.4 Estudios de los efectos sobre las características de los residuos de origen animal. Si el aditivo está destinado a modificar determinadas características de los residuos de origen animal (por ejemplo, nitrógeno, fósforo, olor, volumen) se exigirán estudios que prueben estas propiedades.
4. Capítulo 4: Estudios sobre la seguridad de empleo del aditivo
Los estudios indicados en este capítulo se destinarán a evaluar:
La seguridad de empleo del aditivo para las especies diana,
Los riesgos asociados con la selección y transferencia de la resistencia a los antibióticos y el incremento de la persistencia y excreción de patógenos intestinales,
Los riesgos que pueda representar para el consumidor la ingestión de alimentos que contengan residuos del aditivo o de sus metabolitos,
Los riesgos que puedan entrañar la inhalación y el contacto con las mucosas, los ojos o la piel para las personas que manipulen el aditivo puro o incorporado a las premezclas o a los piensos,
Los riesgos de los efectos negativos para el medio ambiente del propio aditivo o de los productos derivados del mismo, ya sea directamente o a través de los excrementos de animales.
Se tendrán en cuenta las incompatibilidades e interacciones conocidas entre el aditivo y medicamentos veterinarios y componentes de dietas pertinentes para las especies en cuestión.
Normalmente, se requerirá la totalidad de estos estudios para todos los aditivos, a menos que la Directiva contenga exclusiones o modificaciones específicas respecto a alguno.
En general, se aceptará un número más limitado de estudios cuando se proponga ampliar la autorización de empleo a especies que se encuentren fisiológica y metabólicamente próximas a la especie para la cual el empleo del aditivo ya está autorizado. Este conjunto reducido de datos demostrará la seguridad en relación con las nuevas especies, así como la ausencia de diferencias importantes en relación con la evolución metabólica y los residuos en los tejidos comestibles. Se justificará el LMR propuesto y el plazo de retirada relativo a las especies en cuestión.
Con objeto de evaluar los riesgos para el consumidor y establecer en consecuencia los LMR y el plazo de retirada, se proporcionará la siguiente información:
La estructura química de la sustancia activa,
El metabolismo de las especies diana propuestas,
La naturaleza de los residuos en estas especies diana, Estudio de depleción de los tejidos de los residuos,
Datos sobre los efectos biológicos de la sustancia activa y sus metabolitos.
El conocimiento de la biodisponibilidad de los residuos (ligados y no ligados) podrá ser de utilidad especialmente en caso de que se produzcan muchos metabolitos y no se pueda señalar ningún residuo marcador (véase punto 4.1.3.3).
Además, el conocimiento de la composición y de las propiedades fisicoquímicas y biológicas de las principales sustancias en los excrementos derivadas del aditivo es imprescindible para determinar los estudios necesarios para la evaluación del riesgo de persistencia o de efectos negativos sobre el medioambiente (véase punto 4.5).
4.1 Estudios sobre las especies diana.
4.1.1 Pruebas de tolerancia en las especies diana/categorías de animales. El objetivo es determinar el margen de seguridad (es decir, el margen entre la dosis máxima propuesta en los piensos y la dosis que provoque efectos negativos). Sin embargo, un margen de seguridad de un factor de por lo menos diez se considerará suficiente para no requerir ninguna prueba suplementaria. Esta prueba de tolerancia deberá llevarse a cabo en las especies diana/las categorías animales preferiblemente durante todo el período de producción, si bien normalmente se aceptará un período de prueba de un mes. Esto requiere por lo menos la evaluación de signos clínicos y otros parámetros para determinar efectos en la salud de los animales diana. Se incluirá un grupo de control negativo (sin antibióticos, promotores de crecimiento u otras sustancias medicamentosas). En función del perfil toxicológico, también podrán requerirse parámetros adicionales. En esta sección se comunicará asimismo cualquier efecto nocivo detectado durante las pruebas de eficacia.
Siempre que el producto se destine para uso en animales que pueden utilizarse para la cría, deberán llevarse a cabo estudios para identificar posibles deficiencias en la función reproductiva general masculina o femenina o efectos dañinos en la descendencia derivados de la administración del aditivo investigado.
4.1.2 Seguridad microbiológica del aditivo.
4.1.2.1 Todos los estudios harán referencia al más alto nivel de dosis propuesto.
4.1.2.2 Si la sustancia activa presenta actividad antimicrobiana a nivel de utilización en el pienso, se determinará la Concentración Mínima Inhibitoria (CMI) en las bacterias patógenas y no patógenas, endógenas y exógenas apropiadas, de conformidad con procedimientos estandarizados.
4.1.2.3 Pruebas para determinar la capacidad del aditivo para:
Inducir la resistencia cruzada a los antibióticos pertinentes,
Seleccionar cepas bacterianas resistentes, en condiciones de campo, en las especies diana; en caso afirmativo, se estudiarán los mecanismos genéticos de transferencia de los genes resistentes.
4.1.2.4 Pruebas destinadas a determinar el efecto del aditivo:
Sobre determinados patógenos oportunistas del tracto digestivo (por ejemplo enterobacteriaceae, enterococos y clostridia),
En la diseminación o excreción de microorganismos zoonóticos pertinentes, por ejemplo «salmonella spp», «campylobacter spp».
4.1.2.5 En caso de que la sustancia activa demuestre poseer una acción antimicrobiana, se efectuarán estudios de campo para determinar la resistencia bacteriana del aditivo.
4.1.3 Estudios del metabolismo y de los residuos.
4.1.3.1 El objetivo de los estudios es:
Identificar las vías metabólicas de la sustancia activa como base para su evaluación toxicológica,
Identificar los residuos y determinar su cinética en los tejidos y productos comestibles (leche, huevos), Identificar las sustancias excretadas como requisito previo para evaluar su impacto en el medio ambiente.
En ocasiones, por ejemplo en los aditivos derivados por fermentación, será necesario ampliar estos estudios a otras sustancias añadidas o derivadas durante el proceso de fermentación, como en el caso de la presencia de una toxicidad significativa en relación con la del componente o componentes activos del aditivo.
4.1.3.2 Aspectos farmacocinéticos: En la planificación y el diseño experimental de los estudios se debe tener en cuenta la categoría anatómica, fisiológica (edad, tipo, sexo) y zootécnica y las particularidades ambientales de la población diana. En su caso, se tendrá en cuenta la influencia de la microflora del intestino o de la panza, del ciclo enterohepático y de la cecotrofia. El régimen de dosis de las pruebas será el mismo que el que se pretende utilizar y, en caso justificado, incluso un múltiplo de la mencionada dosis. La sustancia activa (incluida la sustancia etiquetada) se incorporará a la alimentación a menos que exista un motivo justificado para no hacerlo.
Se requerirán los siguientes estudios:
Balance metabólico y cinética del plasma/la sangre tras la administración de una sola dosis a fin de evaluar el índice y grado de absorción, distribución y excreción (orina, excrementos, branquias, bilis, aire expirado, leche o huevos),
Identificación de los principales (> 10 por 100) metabolitos en los excrementos; excepto si uno de los metabolitos menos importantes (> 10 por 100) presenta interés toxicológico,
Distribución del material etiquetado en tejidos y productos tras la administración de una sola dosis a animales que ya han alcanzado un equilibrio constante con el aditivo sin etiqueta.
Los estudios mencionados en los puntos 4.1.3.1 y 4.1.3.3 incluirán el indicador de isótopos o métodos pertinentes alternativos.
4.1.3.3 Estudio de los residuos: Identificación de los residuos (compuesto de origen, metabolitos, productos de degradación, residuos ligados3) que representan más de un 10 por 100 del residuo total (excepto si un metabolito de menor importancia presenta interés toxicológico) en los tejidos y productos comestibles (leche, huevos) en equilibrio metabólico, es decir, después de la administración de varias dosis de la sustancia etiquetada; coeficiente del residuo marcador en relación con los residuos totales,
3 Los residuos ligados corresponden a la fracción residual de tejido que no se puede extraer con medios fisicoquímicos o biológicos. Proceden de la unión covalente de un metabolito del compuesto con macromoléculas celulares.
Estudio cinético de los residuos en los tejidos (incluidos, si procede, la leche y los huevos) durante el período de depleción una vez logrado el equilibrio y utilizando el nivel más alto del perfil metabólico propuesto, determinación del tejido diana4 y del residuo marcador,
4 El tejido diana es el tejido comestible seleccionado para evaluar el conjunto de residuos en el animal diana.
Estudio de depleción del residuo marcador de los tejidos diana (incluidos, en su caso, la leche y los huevos) una vez eliminado el aditivo después de una repetida administración conforme a las condiciones de uso propuestas y que sea suficiente para alcanzar el equilibrio constante, con objeto de establecer un plazo de retirada sobre la base del LMR establecido,
El tiempo de espera del aditivo no será inferior al tiempo necesario para que la concentración del residuo marcador determinado en el tejido diana llegue a ser inferior al valor LMR (límite de confianza de 95 por 100). El establecimiento de los momentos temporales espaciados, convenientemente elegidos por referencia a la fase de depleción de la sustancia activa y de sus metabolitos, y estudiar por lo menos cuatro animales por momento temporal en función de las especies (tamaño, variabilidad genética) se considerará requisito mínimo5.
5 Para determinar el tiempo de espera, se sugieren las siguientes cantidades mínimas de animales sanos muestreados en relación con cada matanza o momento temporal:
Reses lactantes: Ocho, incluidos los animales de la segunda lactancia y las lactancias subsiguientes (cuatro reses de alto rendimiento en una fase temprana de lactancia y cuatro reses de bajo rendimiento en una fase tardía de lactancia),
Otros animales de gran tamaño, cuatro por período de muestreo,
Aves de corral, seis por período de muestreo, Aves ponedoras, diez huevos cada unidad de tiempo, Pescados, diez por período de muestreo.
4.2 Estudios con animales de laboratorio. Estos estudios analizarán la sustancia activa por medio de métodos de prueba estandarizados internacionalmente reconocidos de conformidad con las Directrices de la OCDE sobre detalles metodológicos o las disposiciones del Real Decreto 2043/1994 (BPL). Podrá ser necesario realizar estudios adicionales sobre metabolitos particulares producidos por las especies diana, si éstos no aparecen de forma significativa en las especies de prueba en el laboratorio. Además, si existen datos referentes a las personas, éstos deberán considerarse a la hora de decidir los estudios adicionales que deberán llevarse a cabo.
4.2.1 Toxicidad aguda. Se efectuarán estudios de toxicidad aguda por vía oral por lo menos con dos especies mamíferas. Si procede, se sustituirá una de las especies de laboratorio por una especie diana. No será necesario determinar el LD50 exacto; normalmente, se requerirá una determinación aproximada de la dosis letal mínima. A fin de reducir el número de animales necesarios y de minimizar su sufrimiento, la dosis máxima no deberá sobrepasar los 2.000 mg/kg de peso corporal; se recomienda aplicar métodos alternativos (ensayo límite, método de dosis fija, método de clase tóxica aguda).
Los riesgos para los trabajadores se evaluarán mediante una serie de estudios en los que se utilizará el producto (sustancia activa más excipiente en la forma en que se comercializará). Se realizarán estudios sobre el efecto irritante en la piel y, si los resultados son positivos, se evaluará el efecto irritante en las membranas mucosas (por ejemplo, el ojo). Asimismo, se evaluará el potencial alergénico y el potencial de sensibilización cutánea. En caso de que el producto pueda formar polvo o neblina susceptibles de ser respirados, se realizarán estudios sobre la toxicidad aguda por inhalación del producto.
4.2.2 Estudios de genotoxicidad, incluida la mutagenicidad. A fin de identificar sustancias activas y, si procede, sus metabolitos y sus productos de degradación con propiedades mutágenas y genotóxicas, se realizará una combinación selectiva de por lo menos tres pruebas diferentes de genotoxicidad. Normalmente, la batería de ensayos incluirá pruebas de los sistemas procariótico y eucariótico, incluidos los sistemas de ensayo in vitro e in vivo en mamíferos. Si procede, las pruebas se llevarán a cabo con y sin activación metabílica mamífera.
Se justificará la elección de las pruebas en términos de fiabilidad para evaluar los efectos genotóxicos en relación con diversos aspectos genéticos a nivel de gen, cromosoma y genoma. En función de los resultados de las pruebas, del perfil toxicológico general de la sustancia y del uso previsto, podrá indicarse la conveniencia de efectuar exámenes complementarios. Los ensayos se realizarán de conformidad con procedimientos establecidos, actualizados y validados. Si el objetivo del ensayo es la médula ósea, en caso de que el resultado sea negativo se requerirá una prueba de la exposición de las células a la sustancia de ensayo.
4.2.3 Estudios de toxicidad oral subcrónica (noventa días). La duración de los ensayos será, como mínimo, de noventa días. Por lo que se refiere a los aditivos destinados al uso en especies animales utilizadas en la producción de alimentos, los ensayos se llevarán a cabo con dos especies animales, una de las cuales será una especie no roedora, que pueden ser las especies diana. Por lo que se refiere a los aditivos destinados al uso en animales que no se destinan al consumo humano, bastarán los estudios sobre las especies diana: A fin de obtener una reacción a la dosis, la sustancia activa se administrará oralmente por lo menos a tres niveles y a un grupo de control.
Normalmente, la dosis máxima provocará efectos dañinos. La dosis mínima no debería provocar ningún efecto tóxico.
4.2.4 Estudios de toxicidad oral crónica (incluidos los estudios de carcinogenicidad). Los estudios de toxicidad crónica, que podrán incluir el examen de la carcinogenicidad, deberán efectuarse por lo menos con una especie de roedores.
El examen de la carcinogenicidad no será necesario si la sustancia activa y sus metabolitos:
Dan regularmente resultados negativos en una gama adecuada de pruebas de genotoxicidad,
No están estructuralmente relacionados con agentes carcinógenos conocidos, y
No señalan ningún efecto indicativo de una posible (pre)neoplasia en ensayos de toxicidad crónica.
4.2.5 Estudios de toxicidad reproductiva, incluida la teratogenicidad.
4.2.5.1 Estudio de toxicidad reproductiva en dos generaciones: Los estudios de los efectos en la reproducción se extenderán como mínimo a dos generaciones en línea directa (F1, F2) y podrán combinarse con un examen de teratogenicidad. La sustancia objeto del estudio se administrará a machos y hembras en un momento adecuado previo al apareamiento. La administración se prolongará hasta el destete de la segunda generación filial (F2).
Todos los parámetros referentes a la fertilidad, la gestación, el parto, el comportamiento materno, la lactancia, el crecimiento y el desarrollo de la primera generación filial (F1) desde la concepción hasta la madurez, y el desarrollo de la segunda generación filial (F2) hasta el destete deberán estudiarse y anotarse cuidadosamente.
4.2.5.2 Estudio de teratogenicidad: El estudio de teratogenicidad incluye el examen de embriotoxicidad y de fetotoxicidad. Se realizará por lo menos en dos especies.
4.2.6 Estudios de metabolismo y disposición. Se realizarán estudios sobre absorción, distribución en los fluidos y tejidos corporales y vías de excreción. Se llevará a cabo un estudio metabólico, incluidos el equilibrio y la determinación metabólicos de los principales metabolitos en la orina y los excrementos, en animales de ambos sexos de las mismas especies que los utilizados en los estudios toxicológicos. Se administrará una sola dosis de la molécula etiquetada (véase punto 4.1.3) en estado de equilibrio constante alcanzado con el uso del compuesto no etiquetado en una dosis igual al nivel más alto propuesto para el uso en el animal diana.
4.2.7 Biodisponibilidad de los residuos. Para la evaluación del riesgo para los consumidores de determinados residuos de los productos de origen animal, a saber, los residuos ligados, se podrá tener en cuenta un factor de seguridad adicional basado en la determinación de su biodisponibilidad con animales de laboratorio adecuados y métodos reconocidos.
4.2.8 Otros estudios toxicológicos y farmacológicos específicos. Si persiste algún motivo de duda, se llevarán a cabo estudios adicionales que proporcionen información complementaria de utilidad para la evaluación de la seguridad de la sustancia activa y sus residuos.
4.2.9 Determinación del nivel de efecto nulo (NOEL). Para determinar un NOEL expresado en términos de mg/kg de peso corporal diario se tendrán en cuenta todos los resultados anteriormente mencionados, así como todos los datos pertinentes publicados (incluida cualquier información sobre los efectos de la sustancia activa en el ser humano) y, en su caso, información sobre estructuras químicas estrechamente relacionadas. Se seleccionará el NOEL más bajo.
Sin embargo, el NOEL que deberá utilizarse para el cálculo de la IDA se seleccionará sobre la base de los efectos toxicológicos o farmacológicos según el caso. La IDA para determinados aditivos, por ejemplo los antibacterianos, se establecerá sobre la base de los efectos en la microflora del intestino humano. A falta de métodos aceptados y validados internacionalmente para describir la flora del intestino, será más adecuado describir los efectos en cepas bacterianas sensibles del intestino humano seleccionadas.
4.3 Evaluación de la seguridad para el consumo humano.
4.3.1 Propuesta de ingesta diaria admisible (IDA) del aditivo. Si procede, se propondrá un valor IDA.
El valor IDA (expresado en términos de miligramos de aditivo o de material relacionado con el aditivo por persona por día) se deriva dividiendo el NOEL (el nivel de efecto nulo) por un factor de seguridad adecuado y multiplicando por un peso corporal humano medio de 60 kilogramos. Este NOEL expresado en términos de miligramos por kilogramo de peso corporal por día, se podrá seleccionar sobre la base de datos biológicos o farmacológicos disponibles. En algunos casos, el valor IDA basado en las propiedades microbiológicas de los aditivos será más pertinente. La elección dependerá de qué propiedad es la más pertinente en términos de riesgo para la salud del consumidor.
El factor de seguridad utilizado para determinar el valor IDA para un aditivo específico se seleccionará teniendo en cuenta lo siguiente:
La naturaleza del efecto biológico utilizado para identificar el NOEL,
La pertinencia de este efecto sobre el hombre y su reversibilidad,
La gama y calidad de los datos utilizados para identificar el NOEL,
Cualquier conocimiento sobre el efecto de los componentes del residuo.
Para calcular el valor IDA es habitual emplear un factor de seguridad de por lo menos 100 (es decir, un factor 10 a fin de tener en cuenta la posible variación entre especies y otro factor 10 a fin de tener en cuenta las posibles diferencias en las reacciones individuales de los seres humanos). Si se dispone de datos sobre la sustancia activa en relación con el ser humano, se aceptará un factor más bajo de seguridad.
4.3.2 Propuesta de límites máximos de residuos (LMR) del aditivo. Para calcular el LMR se asume que la ingesta de tejidos comestibles, productos de la leche y los huevos forma la única fuente posible de exposición para el ser humano. En caso contrario, deberá solicitarse autorización para otras fuentes.
Varias de estas sustancias se han utilizado tanto como aditivos en la alimentación animal como en otras aplicaciones. En estos casos, los LMR calculados deberían ser idénticos. Puede haber casos en que, estrictamente por consideraciones científicas, se calcule un LMR diferente para cada uso cuando la vía, la cantidad, la frecuencia de dosificación y la duración de la misma difieren lo suficiente de las adecuadas para el empleo del aditivo en los piensos como para suponer que los procesos cinéticos y metabólicos arrojarán un perfil de residuos distinto. En estas circunstancias, se aplicará el LRM más estricto.
Para establecer un LMR se definirá la naturaleza química del material relacionado con el fármaco utilizado para determinar los niveles de residuo en el tejido. Es lo que se denomina residuo marcador, que no ha de ser necesariamente el residuo toxicológicamente pertinente, sino que se debe elegir como indicador conveniente para representar al residuo total significativo. Se establecerán los coeficientes del residuo marcador/los residuos totales en relación con el valor IDA (es decir, el coeficiente del residuo marcador/total de los residuos radiactivos, residuo de marcador/todos los residuos biológicamente activos) para todo el período de duración de los estudios de depleción. En especial, deberá conocerse este coeficiente en relación con el momento de determinación de los LMR. Asimismo, se dispondrá de un método analítico adecuado de este residuo marcador, a fin de asegurar que se respeta el LMR.
Para establecer los LMR (expresados en términos de g/kg de residuo marcador por kilogramo de tejido o producto blando comestible) sobre la base de un valor IDA, se aplicarán los siguientes valores relativos al consumo diario de alimentos para seres humanos:
|
Mamíferos – Gramos |
Aves – Gramos |
Pescado – Gramos |
|
|---|---|---|---|
| Carne. | 300 | 300 | 300 (*) |
| Hígado. | 100 | 100 | |
| Riñones. | 50 | 10 | |
| Grasa. | 50 (**) | 90(***) | |
| + Leche. | 1.500 | ||
| + Huevos. | 100 |
(*) Músculo y piel en proporciones normales.
(**) Para cerdos, 50 g de grasa y piel en proporciones normales.
(***) Grasa y piel en proporciones normales.
Los LMR individuales en diferentes tejidos deberán reflejar la cinética de depleción de los residuos en los tejidos de las especies animales destinados a uso. Se requerirá un método analítico con un límite de cuantificación por debajo del LMR (véase la sección II, punto 2.5.3).
Si una sustancia puede producir residuos en los tejidos y los productos, los LMR propuestos serán tales que la cantidad total de residuo toxicológicamente (o microbiológicamente) significativo injerido diariamente6 esté por debajo del valor IDA (véase el cuadro anterior).
6 Cálculo propuesto: [500 gramos de carne (que puede ser 300 gramos de músculo, 100 gramos de hígado, 50 gramos de riñón, 50 gramos de grasa) o 500 gramos de aves de corral (que puede ser 300 gramos de músculo, 100 gramos de hígado, 10 gramos de riñón, 90 gramos de grasa) o 300 gramos de pescado] + 1.500 gramos de leche + 100 gramos de huevo.
El LMR sólo se establecerá previa consideración e inclusión de otras posibles fuentes de exposición del consumidor a los componentes de los residuos.
Los residuos relacionados con determinados aditivos podrán presentar niveles inferiores a los valores LMR en la leche, los huevos o la carne y, no obstante, incidir en la calidad de los alimentos, especialmente en determinados procesos de transformación de los alimentos, por ejemplo, el empleo de leche en la fabricación de quesos. Para tales aditivos, además del establecimiento de valores LMR, convendrá considerar un «residuo máximo compatible con el proceso de transformación (de productos alimenticios)».
No se requerirá valor LMR en los siguientes casos:
No hay biodisponibilidad de los residuos ni se produce ningún efecto dañino en el intestino humano, incluida su microflora,
Degradación total en nutrientes o en sustancias inofensivas en las especies diana,
«No se especifica» el valor IDA a causa de una toxicidad baja en las pruebas con animales,
En los casos en que se emplee únicamente en la alimentación de animales de compañía,
En los casos en que una sustancia está autorizada como aditivo alimentario7, normalmente no se requerirá LMR si el residuo marcador es fundamentalmente la sustancia madre y constituye sólo una fracción insignificante del IDA del aditivo alimentario.
7 De conformidad con el Real Decreto 1111/1991, de 12 de julio, por el que se modifica la Reglamentación Técnico-Sanitaria de aditivos alimentarios, aprobada por Real Decreto 3177/1983, de 16 de noviembre, y modificada por el Real Decreto 1339/1998, de 28 de octubre.
4.3.3 Propuesta de tiempo de espera para el aditivo. El tiempo de espera se establecerá sobre la base de los niveles LMR. El tiempo de espera comprende el período inmediatamente posterior al cese de la administración de la fórmula del aditivo propuesta, necesario para permitir que los niveles de residuo se sitúen por debajo de los niveles de LMR (límite de confianza del 95 por 100).
Para establecer un plazo de espera, se podrá identificar un tejido comestible particular, a menudo denominado tejido diana, como sustituto de otros.
4.4 Evaluación de la seguridad de los trabajadores. Los trabajadores pueden estar sometidos principalmente a exposición por inhalación o a exposición tópica durante la fabricación, la manipulación o el empleo del aditivo; por ejemplo, los trabajadores agrícolas están potencialmente expuestos durante los procesos de manipulación y mezcla del aditivo. Se proporcionará información adicional relativa al modo como se deben manipular las sustancias. Se incluirá una evaluación del riesgo para los trabajadores.
La experiencia en la fábrica es con frecuencia una fuente importante de información para evaluar los riesgos para los trabajadores derivados de la exposición al propio aditivo, tanto por vía respiratoria como por vía cutánea. Especial atención merecen los aditivos/los piensos tratados con aditivos y los excrementos animales que se presentan en forma de polvo seco, o pueden dar lugar al mismo, así como los aditivos de los piensos que pueden tener potencial alergénico.
4.4.1 Evaluación del riesgo toxicológico en relación con la seguridad de los trabajadores.
4.4.1.1 Efectos en el sistema respiratorio: Se proporcionarán pruebas de que los niveles de polvo en el aire no son peligrosos para la salud de los trabajadores. En caso necesario, se incluirán las siguientes pruebas:
Pruebas de inhalación en animales de laboratorio, datos epidemiológicos publicados y los datos del solicitante en relación con su fábrica y con las pruebas de irritabilidad y sensibilización del sistema respiratorio.
4.4.1.2 Efectos en los ojos y la piel: Si se tienen, se proporcionarán pruebas directas de la ausencia de irritabilidad y sensibilización en las personas en determinadas situaciones, que se completarán con los resultados de pruebas validadas en animales en relación con la irritación de la piel y el ojo, y con el potencial de sensibilización, efectuadas con el aditivo adecuado.
4.4.1.3 Toxicidad sistémica: Los datos sobre toxicidad derivados del cumplimiento de los requisitos de seguridad (incluidas la toxicidad por administración repetida, la mutagenicidad, la carcinogenicidad y la prueba de reproducción) se utilizarán para evaluar otros aspectos de la seguridad de los trabajadores, para lo que se tendrá en cuenta que la contaminación cutánea y la inhalación del aditivo constituyen las vías de exposición más probables.
4.4.2 Evaluación de la exposición. Se proporcionará información sobre el modo de empleo del aditivo que probablemente causa exposición a través de todas las vías, es decir, por inhalación, por ingestión o por vía cutánea. Esta información incluirá, a ser posible, una evaluación cuantitativa, como la concentración típica en la atmósfera, la contaminación cutánea o la ingestión. En caso de que no se disponga de información cuantitativa, se proporcionará información suficiente para permitir una evaluación adecuada de la exposición.
4.4.3 Medidas de control de la exposición. De la información de la evaluación toxicológica y de exposición se extraerá una conclusión sobre los riesgos para la salud de los usuarios (sistémicos, de toxicidad, irritación o sensibilización) al aplicar las medidas de control de la exposición razonables dadas las circunstancias. Si el riesgo es inaceptable, se introducirán medidas de precaución a fin de controlar o eliminar el riesgo de exposición. La mejor solución es la reformulación o la modificación del proceso de producción, empleo o eliminación del aditivo. El uso de equipos de protección individual se considerará únicamente como último recurso de protección para proteger contra cualquier riesgo residual una vez se hayan introducido las medidas de control.
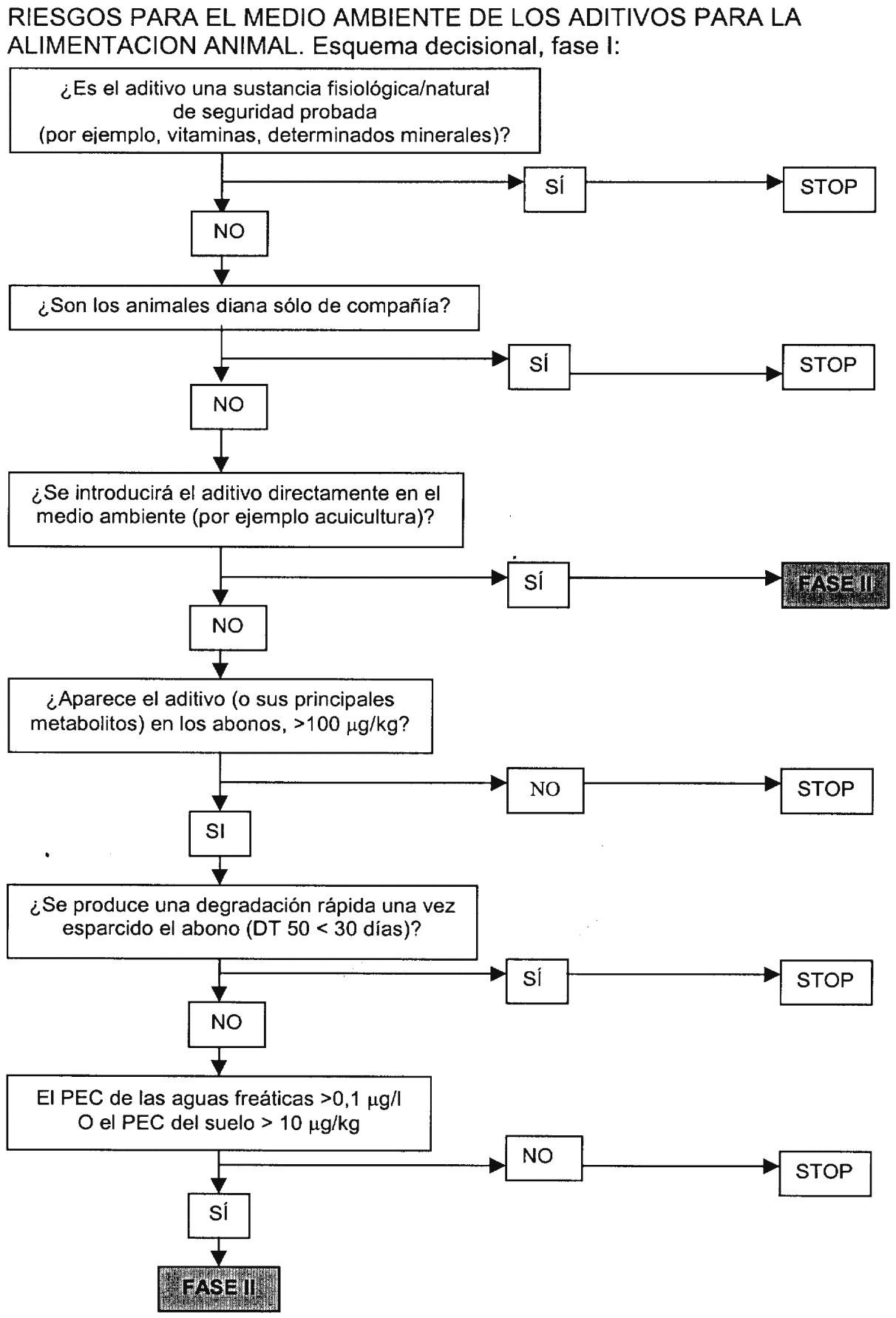
4.5 Evaluación del riesgo para el medio ambiente. Es importante tener en cuenta las consecuencias para el medio ambiente de los aditivos en los piensos, puesto que la administración de aditivos en la alimentación animal se realiza normalmente durante un largo período de tiempo (incluso durante toda una vida), puede afectar a numerosos grupos de animales y muchos aditivos se absorben mal y aparecen prácticamente intactos en los excrementos. No obstante, en algunos casos, la necesidad de evaluar los riesgos para el medio ambiente será limitada. No resulta adecuado establecer pautas estrictas en las presentes directrices generales. Para determinar el impacto medioambiental de un aditivo en la alimentación animal, se adoptará un planteamiento gradual (véase esquema decisional), en el que, en una primera fase, se identifiquen claramente los aditivos que no necesitan más pruebas. Para los demás aditivos será necesario introducir una segunda fase de pruebas (fase II A) a fin de obtener información adicional sobre la base de la cuál se podrá considerar necesario llevar a cabo estudios ulteriores (fase II B). En su caso, los estudios se llevarán a cabo de conformidad con las disposiciones del Real Decreto 2043/1994.
4.5.1 Fase I de evaluación. El objetivo de la fase I de evaluación es determinar, en gran parte sobre la base de datos ya establecidos para otros fines, si es probable que un aditivo o sus metabolitos presenten efectos ambientales significativos.
No se realizará la fase II de evaluación sobre la base de uno de los dos criterios siguientes:
a) La naturaleza química y el efecto biológico del aditivo y de su uso indican que el impacto será insignificante: es decir, en los casos en que el aditivo o su metabolito o metabolitos principales (más del 20 por 100 del total de residuos en los excrementos) son:
Sustancias fisiológicas/naturales (por ejemplo una vitamina o un mineral) que no alterarán la concentración en el ambiente, a menos que exista un motivo evidente que aconseje la evaluación (por ejemplo, cobre),
Aditivos destinados a los animales de compañía (excluidos los caballos).
b) En el peor de los casos, la concentración ambiental prevista (PEC) es demasiado baja para resultar peligrosa.
Probablemente, el peor valor PEC de suelo aparecerá al esparcir en la tierra el abono generado durante el período de máxima excreción de los constituyentes residuales más importantes (el aditivo y/o sus principales metabolitos). Deberá evaluarse el valor PEC en relación con cada uno de los principales componentes residuales en el abono y en relación con cada compartimiento en cuestión. Por lo que se refiere al compartimiento terrestre, no se requerirá ninguna evaluación adicional si el valor PEC no excede de los 100 µg/kg para el conjunto de los principales componentes residuales en el abono, o si los principales componentes residuales en el abono ya se han convertido (tiempo de degradación DT 50 < treinta días) (en caso de que estos datos estén disponibles) en componentes naturales o en concentraciones de menos de 100 µg/kg, o si el valor PEC del suelo (5 centímetros de profundidad) es menor de 10 µg/kg.
Por lo que se refiere al agua, el peor valor PEC aparecerá ya sea por vertido directo de los piensos o los excrementos que contienen el aditivo y sus metabolitos en depósitos de agua o por medio de lixiviación del material que se encuentra en los excrementos o en el suelo en las aguas freáticas. Si se puede establecer de forma admisiblemente fiable que el valor PEC relativo a la contaminación de los depósitos de agua o del agua freática es menor de 0,1 µg por litro, no será necesario llevar a cabo la fase II de la evaluación del efecto medioambiental del aditivo en los medios acuáticos.
En general, si el solicitante no puede demostrar que el aditivo se puede clasificar en una de estas categorías de exención, o si el aditivo se libera directamente en el medio ambiente (por ejemplo, en la acuicultura), se requerirá la fase II de la evaluación.
4.5.2 Fase II de la evaluación. La fase II de la evaluación se realizará en dos partes: fase II A y fase II B. Se evaluará el potencial de bioacumulación del aditivo y de sus principales metabolitos, así como su influencia sobre el margen de seguridad previsto. Se considera que la bioacumulación no es potencialmente significativa si, por ejemplo, el coeficiente de reparto, Kow, es < 3. Generalmente se requerirán las pruebas adecuadas de la fase II si no se pueden establecer estos márgenes de seguridad.
4.5.2.1 Fase II A: El objetivo de la fase II A de la evaluación es identificar el riesgo para el medio ambiente por medio de:
El refinamiento del cálculo de los valores PEC;
La determinación de la relación entre la exposición, los niveles del aditivo y/o de sus principales metabolitos y los efectos nocivos a corto plazo en especies animales y vegetales sustitutas pertinentes para el compartimiento ambiental objeto de estudio;
El uso de estos resultados para determinar los valores de la concentración prevista sin efecto (PNEC).
Para determinar el riesgo se recomienda el siguiente procedimiento secuencial:
a) Si no se completa en la fase I, se calculará un valor PEC más preciso para cada compartimiento ambiental objeto de estudio. Para determinar el valor PEC se tendrá en cuenta lo siguiente:
La concentración de aditivo y/o de sus principales metabolitos en el abono tras la administración a animales del nivel de dosis propuesto del aditivo. Este cálculo tendrá en cuenta el volumen de excrementos y los índices de dosificación;
La posible dilución del material excretado relacionado con el aditivo por proceso normal de fabricación y almacenamiento del abono antes de ser utilizado en la tierra; La adsorción/desorción del aditivo y de sus metabolitos en el suelo, la persistencia de residuos en el suelo (DT50 y DT90); el sedimento en el caso de la acuicultura;
Otros factores como la fotolisis, la hidrólisis, la evaporación, la degradación en sistemas de sedimentación en el suelo o en el agua, la dilución por medio del arado, etcétera.
A efectos de la fase II A de la evaluación del riesgo se adoptará el valor PEC más alto obtenido con estos cálculos en relación con cada compartimiento ambiental objeto de estudio.
En caso de preverse una alta persistencia en el suelo (DT90 > un año) de concentraciones superiores a 10 g/kg de suelo en equilibrio constante, podrá ser necesario realizar la evaluación prevista en la fase II B.
b) A continuación, se determinarán los niveles en los que aparecen importantes efectos negativos a corto plazo en relación con varios niveles tróficos en los compartimientos ambientales objeto de estudio (suelo, agua). Estas pruebas se realizarán conforme a las directrices de la OCDE8 o a otras directrices similares establecidas. Pruebas pertinentes para el medio ambiente terrestre incluyen: toxicidad para las lombrices de tierra (concentración letal del 50 por 100, valor CL50), fitotoxicidad (concentración efectiva del 50 por 100, valor CE50) en plantas terrestres, efectos sobre los microorganismos del suelo (por ejemplo, CE50 para los efectos de metanogénesis y fijación del nitrógeno). Con respecto al entorno acuático: peces, un estudio de CL50 de noventa y seis horas; «Daphnia magna», un estudio de CE50 de cuarenta y ocho horas; algas, un estudio de CL50 y un estudio de toxicidad para los organismos de sedimentación.
8 OECD Guidelines for Testing of Chemicals.
c) Se calculará el valor PNEC para cada compartimiento ambiental objeto de estudio. Normalmente, este cálculo se realizará a partir del valor más bajo observado (es decir, el resultado en la especie más sensible) con un efecto nocivo negativo en las mencionadas pruebas de ecotoxicidad, dividiendo por un factor de seguridad de por lo menos 100 en función del indicador y del número de especies de prueba que se utilicen.
d) Se compararán los valores PEC y PNEC calculados. El coeficiente aceptable del valor PEC con respecto al valor PNEC dependerá de la naturaleza del resultado de la prueba utilizado para determinar el PNEC. Normalmente estará entre 1 y 0,1. Si se identifican coeficientes perceptiblemente más bajos que éstos, es poco probable que sea necesario realizar pruebas ecotoxicológicas adicionales, a menos que se espere una bioacumulación. En cambio, unos coeficientes más altos indicarán la necesidad de realizar pruebas correspondientes a la fase II B de la evaluación.
4.5.2.2 Fase II B (estudios toxicológicos más detallados): Por lo que se refiere a los aditivos en relación a los cuales persisten dudas sobre su impacto medioambiental una vez realizada la fase II A de la evaluación, se requerirán estudios más detallados de sus efectos en las especies biológicas de los compartimientos ambientales que, de acuerdo con los resultados de los estudios de la fase II A, siguen presentando dudas. En este caso, será necesario realizar pruebas adicionales a fin de determinar los efectos crónicos y más específicos en las especies animales, vegetales y microbianas pertinentes. Es posible que en la fase II A de la evaluación se haya sobrestimado el valor PEC; para demostrarlo podrá ser necesario realizar mediciones de las concentraciones ambientales y de la persistencia del aditivo y/o de sus principales metabolitos en situaciones reales de uso.
En varias publicaciones, por ejemplo en las directrices de la OCDE, se describen las pruebas adicionales de ecotoxicidad que conviene realizar. Será necesario considerar tres categorías de especies ambientales, a saber, animales, plantas y microorganismos. Habrá que elegir cuidadosamente estas pruebas, asegurándose de que son las adecuadas para las condiciones de liberación y dispersión del aditivo o sus metabolitos en el ambiente.
La evaluación del impacto en el compartimiento terrestre incluirá:
Un estudio subletal de los efectos en las lombrices de tierra, estudios adicionales sobre el impacto en la microflora del suelo, pruebas de fitotoxicidad en una gama de especies vegetales económicamente importantes, estudios en los invertebrados de los pastos, incluidos insectos y pájaros silvestres.
N.B. Puede que no sea necesario llevar a cabo una evaluación separada de la toxicidad en los mamíferos, ya que, probablemente, este aspecto se evalúe en las pruebas de toxicidad en mamíferos para la determinación del valor IDA.
La evaluación del impacto en el compartimiento acuático incluirá:
Pruebas de toxicidad crónica en los organismos acuáticos más sensibles identificados en la fase II A de la evaluación, por ejemplo: el ensayo en las primeras fases de la vida de los peces, el ensayo de reproducción en Dafnia, pruebas de setenta y dos horas en algas y un estudio de bioacumulación.
En caso de que no se pueda establecer un margen de seguridad adecuado entre los valores PEC y PNEC, se proporcionarán medidas atenuantes eficaces para limitar las consecuencias en el medio ambiente.
5. Capítulo V: Modelo de monografía
5.1 Identidad del aditivo.
5.1.1 Denominación o denominaciones comerciales propuestas.
5.1.2 Tipo de aditivo según su función principal. Se especificará cualquier otro empleo del aditivo.
5.1.3 Composición cualitativa y cuantitativa (sustancia activa, otros componentes, impurezas, variación entre lotes). Si la sustancia activa es una mezcla de componentes activos, cada uno de los cuales es claramente definible, deberán describirse por separado los principales, indicándose su proporción en la mezcla.
5.1.4 Estado físico, distribución granulométrica, forma de las partículas, densidad, densidad aparente; para los líquidos: viscosidad, tensión superficial.
5.1.5 Procedimiento de fabricación, incluidos los posibles tratamientos específicos.
5.2 Características de la sustancia activa.
5.2.1 Denominación genérica, denominación química según la nomenclatura UIQPA, otras denominaciones genéricas internacionales y abreviaturas. Número CAS (Chemical Abstract Service Number).
5.2.2 Fórmula empírica, fórmula estructural y peso molecular. Si la sustancia activa es un producto de fermentación, composición cuantitativa y cualitativa de los principales componentes, origen microbiano (nombre y lugar de la colección de cultivos donde se encuentre depositada la cepa).
5.2.3 Pureza. Composición cuantitativa y cualitativa de las sustancias activas y de las impurezas y las sustancias tóxicas relacionadas, confirmación de la ausencia de organismos de producción.
5.2.4 Propiedades pertinentes. Propiedades físicas de las sustancias químicamente definidas: constante de disociación, pKa, propiedades electrostáticas, punto de fusión, punto de ebullición, densidad, tensión de vapor, solubilidad en agua y en disolventes orgánicos, Kow y Koc, espectro de masa y de absorción, datos sobre los LMR, posibles isómeros y cualquier otra propiedad física pertinente.
5.3 Propiedades fisicoquímicas, tecnológicas y biológicas del aditivo.
5.3.1 Estabilidad del aditivo respecto a agentes atmosféricos como la luz, la temperatura, el pH, la humedad y el oxígeno. Propuesta de período de validez.
5.3.2 Estabilidad durante la preparación de las premezclas y los piensos, en particular frente a las condiciones operacionales previstas (el calor, la humedad, la persistencia de deplazamiento/rozamiento y tiempo). Eventuales productos de degradación o descomposición.
5.3.3 Estabilidad durante la conservación de las premezclas y los piensos en condiciones determinadas. Propuesta de período de validez.
5.3.4 Otras propiedades fisicoquímicas, tecnológicas y biológicas pertinentes, como la capacidad de dispersión en condiciones favorables a fin de formar y conservar mezclas homogéneas en las premezclas y en los piensos, propiedades antiestáticas y en relación con la formación de polvo y capacidad de dispersión en medios líquidos.
5.4 Métodos de control.
5.4.1 Descripción de los métodos aplicados para establecer los criterios enunciados en los puntos 2.1.3, 2.1.4, 2.2.3, 2.2.4, 2.3.1, 2.3.2, 2.3.3 y 2.3.4.
5.4.2 Descripción de los métodos de análisis cualitativo y cuantitativo empleados para determinar el residuo marcador de la sustancia activa en los tejidos y los productos animales diana.
5.4.3 Si dichos métodos han sido publicados, bastará con indicar las referencias bibliográficas, debiéndose facilitar una separata de la publicación.
5.4.4 Información relativa a las condiciones óptimas de conservación para la normativa de referencia.
5.5 Propiedades biológicas del aditivo.
5.5.1 Datos sobre los efectos profilácticos de los coccidiostáticos y otros medicamentos (por ejemplo, morbilidad, mortalidad, número de ooquistes y calificación de las lesiones).
5.5.2 Con respecto a los aditivos zootécnicos distintos de los contemplados en el punto 5.5.1, datos sobre los efectos en la alimentación, el crecimiento, la eficacia nutritiva, la calidad y el rendimiento del producto y cualquier otro parámetro que tenga efectos positivos para el bienestar animal, el medio ambiente, el productor o los consumidores.
5.5.2 Con respecto a los aditivos tecnológicos, los efectos tecnológicos pertinentes.
5.5.4 Cualquier efecto negativo, contraindicación o advertencia (animales diana, consumidores, medio ambiente), incluidas las incompatibilidades biológicas, con detalles sobre su justificación. Se especificará cualquier valor IDA o LMR establecido en relación con otros usos de la sustancia activa.
5.6 Indicación cualitativa y cuantitativa de eventuales residuos encontrados en productos de origen animal después de emplear el aditivo de las condiciones de empleo previstas.
5.7 Si procede, se proporcionarán el valor IDA, los LMR establecidos y el plazo de retirada.
5.8 Otras características pertinentes para la identificación del aditivo.
5.9 Condiciones de uso.
5.10 Fecha.
6. Capítulo VI: Modelo de ficha descriptiva
1. Identidad del aditivo.
1.1 Tipo de aditivo.
1.2 Estado físico.
1.3 Composición cualitativa y cuantitativa.
1.4 Método de análisis del aditivo y los residuos.
1.5 Número de registro comunitario (número CE).
1.6 Envasado.
2. Características relativas a la sustancia activa.
2.1 Nombre genérico, denominación química, número CAS.
Nombre genérico.
Denominación química (IUPAC). Número CAS.
2.2 Fórmula empírica.
3. Propiedades fisicoquímicas, tecnológicas y biológicas del aditivo.
3.1 Estabilidad del aditivo.
3.2 Estabilidad durante la preparación de premezclas y piensos.
3.3 Estabilidad durante la conservación de premezclas y piensos.
3.4 Otras propiedades.
4. Condiciones de uso.
4.1 Especie o categoría de animales, edad máxima si procede.
4.2 Contenido mínimo y máximo en los piensos.
4.3 Contraindicaciones, incompatibilidades.
4.4 Advertencias.
5. Persona responsable de su puesta en circulación.
5.1 Nombre.
5.2 Dirección.
5.3 Número de registro.
6. Fabricante.
6.1 Nombre.
6.2 Dirección.
6.3 Número de autorización o de registro del establecimiento o del intermediario.
7. Fecha.
7. Capítulo VII: Renovación de la autorización de los aditivos cuya autorización está vinculada a un responsable de su puesta en circulación
1. Generalidades. Se elaborarán un expediente y una monografía actualizados de conformidad con las directrices más actualizadas y se proporcionará una lista de todas las variaciones de cualquier tipo que hayan tenido lugar desde la concesión de la autorización para la puesta en circulación o desde la última renovación de la misma.
Se confirmará la adaptación de la monografía y el fichero de seguridad a fin de incluir toda nueva información pertinente para el aditivo o que se requiere actualmente a consecuencia de los cambios en las directrices.
Asimismo, se proporcionará información sobre la validez internacional de la autorización y sobre el volumen de ventas.
2. Identidad de la sustancia activa y del aditivo. Se presentarán pruebas de que no se han introducido modificaciones ni alteraciones en la composición, pureza o actividad del aditivo autorizado. Se comunicará cualquier modificación del proceso de fabricación.
3. Eficacia. Se presentarán pruebas de que, al solicitar la renovación de la autorización, el aditivo mantiene la eficacia declarada bajo condiciones normales de la cría de animales en la Unión Europea, incluido un informe de la experiencia general adquirida con el uso del aditivo y la evaluación del rendimiento.
4. Microbiología. Se prestará especial atención al posible desarrollo de la resistencia a los agentes antimicrobianos en relación con el uso a largo plazo en condiciones reales de empleo. Las pruebas se realizarán sobre el terreno en explotaciones ganaderas, en las que se habrá utilizado regularmente el aditivo durante el mayor tiempo posible. Como organismos de ensayo se utilizará una selección de bacterias intestinales comunes, incluidas las bacterias endógenas y exógenas gram-positivas y gram-negativas pertinentes.
Si las pruebas revelan un cambio en la pauta de resistencia con respecto a los datos originales, se examinará la resistencia cruzada de las bacterias resistentes a los antibióticos pertinentes utilizados para el tratamiento de enfermedades infecciosas en el hombre y los animales. Los más importantes son los antibióticos pertenecientes al mismo grupo que el aditivo, aunque se incluirán también antibióticos de otros grupos en las pruebas.
Se comunicarán los resultados de los programas de supervisión adecuados.
5. Seguridad. Se presentarán pruebas de que, a la luz de los actuales conocimientos, el aditivo sigue siendo seguro bajo las condiciones aprobadas para las especies diana, los consumidores, los manipuladores y el medio el ambiente. Se requerirá una actualización de los requisitos de seguridad relativos al período transcurrido desde la autorización de puesta en circulación o desde la última renovación de la misma, que contendrá la siguiente información:
Informes sobre efectos nocivos, incluidos los accidentes (efectos previamente desconocidos, efectos graves de todo tipo, aumento de la incidencia de los efectos conocidos) en relación con los animales diana, los manipuladores y el medio ambiente. En el informe sobre los efectos nocivos se indicará la naturaleza del efecto, el número de individuos/organismos afectados, el resultado, las condiciones de uso, la evaluación de la causalidad,
Informes sobre incompatibilidades y contaminaciones cruzadas previamente desconocidas,
Información relativa a la evaluación de los residuos si procede,
Cualquier otra información relativa a la seguridad del aditivo.
En caso de no presentar información adicional sobre ninguno de estos factores, deberán identificarse claramente los motivos de su omisión.
8. Capítulo VIII: Nueva solicitud basada en la primera autorización de un aditivo cuya autorización está vinculada a un responsable de su puesta en circulación
Puesto que se podrán utilizar todos o parte de los resultados de la evaluación que haya dado lugar a la autorización inicial, los expedientes elaborados de conformidad con el apartado 3 del artículo 9 quater sólo deberán cumplir los siguientes requisitos:
A estos efectos, se considerará que un aditivo es idéntico si la composición cualitativa y cuantitativa y la pureza de sus componentes activos e inactivos son esencialmente similares, la preparación es la misma y las condiciones de uso son idénticas.
Normalmente, para estos productos no será necesario repetir el análisis farmacológico y toxicológico ni las pruebas de eficacia, sino que podrá presentarse una solicitud abreviada, incluidos los informes de los expertos.
Se presentará toda la información requerida en el capítulo II, así como una monografía completa.
Se proporcionarán datos de los que se derive que las características físicas y químicas del aditivo son esencialmente iguales que las del producto autorizado.
Se requerirá confirmación de que los conocimientos científicos adicionales de la documentación existente sobre el aditivo no han conducido a modificar la evaluación inicial sobre su eficacia desde la autorización de puesta en circulación del aditivo original.
Se prestará especial atención al posible desarrollo de la resistencia a los agentes antimicrobianos en relación con el uso a largo plazo de la sustancia activa en condiciones reales de empleo. Las pruebas se realizarán sobre el terreno en explotaciones ganaderas, en las que se habrá utilizado regularmente la sustancia activa durante el mayor tiempo posible. Como organismos de ensayo se utilizará una selección de bacterias intestinales comunes, incluidas las bacterias endógenas y exógenas grampositivas y gram-negativas pertinentes.
Si las pruebas revelan un cambio en la pauta de resistencia con respecto a los datos originales, se examinará la resistencia cruzada de las bacterias resistentes a los antibióticos pertinentes utilizados para el tratamiento de enfermedades infecciosas en el hombre y los animales. Los más importantes son los antibióticos pertenecientes al mismo grupo que el aditivo, aunque se incluirán también antibióticos de otros grupos en las pruebas.
Se presentarán pruebas de que, a la luz de los actuales conocimientos científicos disponibles, el aditivo sigue siendo seguro bajo las condiciones aprobadas para las especies diana, los consumidores, los manipuladores y el medio ambiente.
Se establecerá la conformidad del tiempo de espera con el LMR.
Parte II. Microorganismos y enzimas9
9 Véanse el Real Decreto 1329/1995, de 28 de julio, por el que se fijan las líneas directrices para la evaluación de los aditivos en la alimentación animal; el Real Decreto 2599/1998, de 4 de diciembre, sobre aditivos en la alimentación de los animales; la Orden de 4 de julio de 1994 sobre utilización y comercialización de enzimas, microorganismos y sus preparados en la alimentación animal, y la Orden de 6 de noviembre de 1995 por la que se modifica el anexo del Real Decreto 1329/1995, por el que se fijan las líneas directrices para la evaluación de los aditivos en la alimentación animal.
Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid




