State Agency Official State Gazette






Content not available in English
Excelentísimos señores:
El elevado coste de algunas de las materias primas que intervienen en la fabricación de productos fitosanitarios, fertilizantes y materias afines, así como diversas causas derivadas de su comercialización, distribución y utilización, determinan el hecho de que se vengan empleando como productos fitosanitarios y fertilizantes distintas materias que, en algunos casos, no cumplen las disposiciones legales vigentes, por lo cual se hace imprescindible el control analítico de sus diferentes componentes
Por otra parte, la actuación de los Organismos oficiales en el ámbito de sus respectivas competencias requiere realizar el análisis de los mismos componentes.
Por todo lo anterior, parece lógico unificar criterios y aunar esfuerzos mediante el establecimiento de Métodos de Análisis Oficiales únicos para todos los Ministerios afectados.
Para la redacción de los citados métodos de análisis, hecha conjuntamente por cualificados especialistas de los Ministerios interesados, se ha considerado conveniente adaptarse en lo posible a los aprobados por Organismos internacionales especializados en la materia, con el fin de aprovechar la experiencia habida en su aplicación y de facilitar la confrontación de los resultados en las relaciones comerciales supranacionales.
En su virtud, a propuesta de los Ministerios de Agricultura, Industria y Hacienda, esta Presidencia del Gobierno dispone:
Primero. Se aprueban como oficiales los métodos de análisis de productos fitosanitarios y de fertilizantes que se citan en los anexos 1 y 2.
Segundo. Cuando no existan métodos oficiales para determinados análisis, y hasta que sean estudiados por el Grupo de Trabajo correspondiente, podrán ser utilizados los adoptados por Organismos nacionales o internacionales de reconocida solvencia.
Tercero. Quedan derogadas las disposiciones de igual o inferior rango que se opongan a la presente Orden.
Cuarto. La presente disposición entrará en vigor a los treinta días de su publicación en el «Boletín Oficial del Estado».
Lo digo a VV. EE. a los procedentes efectos.
Dios guarde a VV. EE.
Madrid, 30 de noviembre de 1976.
OSORIO
Excmos. Sres. Ministros de Agricultura, Industria y Hacienda.
METODOS DE ANALISIS DE PRODUCTOS FITOSANITARIOS
Métodos químicos
1. FENITROTION
1.1. Principio.
El método se basa en la medida espectrofotométrica de la absorción a 271 nm. después de efectuar una purificación del principio activo por cromatografía en columna.
1.2. Material y aparatos.
1.2.1. Espectrofotómetro ultravioleta.
1.2.2. Columna para cromatografía de 40 x 0,8 cm.
1.3. Reactivos.
1.3.1. Mezcla n-Hexano-cloroformo:
Mezclar dos partes de n-Hexano con una parte de cloroformo.
1.4. Procedimiento.
Preparación de la columna:
Mezclar 8 g. de sílica gel para cromatografía, empleando 5 ml. de la mezcla n-Hexano-cloroformo y añadirla a la columna.
Pesar una cantidad de muestra que contenga aproximadamente 500 mg. y diluir hasta 50 ml. con la mezcla de n-Hexano-cloroformo. Tomar exactamente 5 ml. de esta solución y llevar a la columna de cromatografía.
A continuación lavar la columna con 70 ml. de la mezcla n-Hexano-cloroformo, inmediatamente después añadir la cantidad necesaria de cloroformo para poder recoger exactamente 50 ml., tomar 2 ml. y llevar a un matraz aforado de 100 ml., diluyendo nuevamente con cloroformo hasta el enrase. Medir la absorbancia a 271 nm. usando cloroformo como referencia.
1.5. Cálculos.
La cantidad de fenitrotión se obtiene de la curva de calibración absorbancia/concentración de fenitrotión puro.

p' = peso, en gramos, de fenitrotión.
p = peso, en gramos, de la muestra.
1.6. Observaciones.
Para pasar a g/l. multiplicar por 10 y por la densidad a 20 °C/4 °C.
2. 2,4 DICLOROFENOXIACETICO SAL AMINA
2.1. Principio.
Por acidificación de la muestra se separa el ácido 2,4-D que es extraído con éter etílico, solución en la que una vez lavada, se determina el contenido en ácido 2,4-D por valoración directa con hidróxido sódico 0,1 N. Posteriormente se determinará el punto de fusión del ácido 2,4-D obtenido según el método físico correspondiente.
2.2. Material y aparatos.
2.2.1. Matraces erlenmeyer de 250 ml.
2.2.2. Embudos de decantación de 250 ml.
2.2.3. Probeta de 100 ml.
2.2.4. Probeta de 50 ml.
2.2.5. Probeta de 10 ml.
2.2.6. Bureta de 50 ml. con divisiones de 1/10 ml.
2.3. Reactivos.
2.3.1. Acido clorhídrico (d = 1,10).
2.3.2. Éter etílico exento de acidez.
2.3.3. Etanol (alcohol etílico neutro 95 por 100).
2.3.4. Hidróxido sódico 0,1 N.
2.3.5. Rojo de metilo (solución alcohólica/acuosa 0,1 por 100).
2.4. Procedimiento.
Pesar con precisión una cantidad de muestra que contenga del orden de 1 g. de ácido 2,4-D en un matraz erlenmeyer de 250 ml., diluirla con agua destilada hasta un volumen de unos 80 ml. y traspasarla a un embudo de decantación de 250 ml., empleando un volumen de unos 20 ml. de agua destilada para hacer el traspaso cuantitativo. Adicionar 3 ml. de ácido clorhídrico y extraer tres veces con 25 ml. de éter etílico cada vez, dejando a cada decantación un tiempo de reposo de 15 minutos.
El conjunto de los extractos etéreos se lava tres veces con 10 ml. de agua destilada (1) cada vez, dejando el tiempo de decantación necesario para tener una separación total de las fases (2). Al final de estos lavados comprobar que el pH de la última fase acuosa separada es aproximadamente 7. Transvasar la fase etérea a un matraz erlenmeyer de 250 ml. Pasar las aguas de lavado al embudo de decantación y efectuar una extracción con 30 ml. de éter etílico, dejando Un tiempo de decantación de 1 hora. Descartar la fase acuosa, lavar dos veces la fase etérea con 25 ml. de agua destilada cada vez (1), dejando un tiempo de decantación de 30 minutos y comprobando que su pH es aproximadamente 7. Reunir esta fase etérea con la anterior situada en el matraz erlenmeyer.
(1) No emplear mayor cantidad de agua que la indicada. Para prevenir la formación de emulsiones puede emplearse en el primer lavado una solución acuosa de cloruro sodio 10 por 100.
(2) Caso de formarse una interfase estable, separarla con la fase acuosa, que posteriormente será de nuevo extraída.
Adicionar a la fase etérea 60 ml. de etanol y valorar con hidróxido sódico 0,1 N., utilizando rojo de metilo como indicador.
Realizar de forma análoga un ensayo en blanco, empleando 9 ml. de etanol y 40 ml. de agua destilada recientemente hervida.
2.5. Cálculos.

N = normalidad de la solución de NaOH.
a = volumen, en ml., de hidróxido sódico 0,1 N utilizado para la muestra.
b = volumen, en ml., de hidróxido sódico 0,1 N utilizado para el ensayo en blanco. p = peso, en gramos, de la muestra1.
d204 = densidad a 20/4 °C.
2.6. Referencias.
1. «Specifications for Pesticides», Who, Geneva, 1961, página 364.
2. «Cipac Handbcok», CIPACL, Harpenden, 1970, pág. 261.
3. 2,4 DICLOROFENOXIACETICO ESTER
3.1. Principio.
El éster contenido en la formulación se hidroliza con hidróxido de litio, la solución resultante se extrae con éter etílico y la fase etérea, previamente lavada con agua, se descarta. La fase acuosa se acidifica con ácido clorhídrido y los ácidos se extraen con éter etílico; la fase etérea se lava con agua hasta reacción neutra y las aguas de lavado son nuevamente extraídas con éter etílico. En el conjunto de los extractos etéreos, se determina el ácido 2,4-D por valoración con hidróxido sódico en solución de etanol. Posteriormente se determinará el punto d fusión del ácido 2,4-D obtenido según el método físico correspondiente.
3.2. Material y aparatos.
3.2.1. Matraces erlenmeyer esmerilados de 250 ml.
3.2.2. Refrigerante Dimroth para reflujo.
3.2.3. Embudos decantación de 250 ml.
3.2.4. Embudo rama corta de 5 cm. de diámetro.
3.2.5. Probeta de 50 ml.
3.2.6. Bureta de 50 ml. con divisiones de 1/10 ml.
3.2.7. Probeta de 10 ml.
3.2.8. Matraz aforado de 200 ml.
3.2.9. Pipeta de 100 ml. con doble enrase.
3.2.10. Probeta de 25 ml.
3.2.11. Agitador magnético provisto de plato calentable.
3.2.12. Bureta de 50 ml. en divisiones de 1/10 ml.
3.3. Reactivos.
3.3.1. Hidróxido de litio.
3.3.2. Éter etílico exento de acidez.
3.3.3. Agua destilada.
3.3.4. Acido clorhídrido (d = 1,19).
3.3.5. Etanol (alcohol etílico neutro 95 por 100).
3.3.6. Hidróxido sódico.
3.3.7. Azul de bromotimol (solución acuosa 0,2 por 100).
3.3.8. Papel indicador de pH universal.
3.4. Procedimiento.
Pesar, con precisión, una cantidad de muestra que contenga 2 g. de ácido 2,4-D. En un matraz erlenmeyer de 250 ml. esmerilado, adicionar 10 ml. de etanol y 25 ml. de solución de. hidróxido de litio 1 N. Colocar el refrigerante y calentar con agitación durante una hora a reflujo intenso. Enfriar y transvasar, con agua destilada, a un embudo de decantación de 250 ml.; el volumen final será de unos 80 ml.
Extraer con 30 ml., de éter etílico, dejando un tiempo de decantación de 30 minutos (hasta obtener una separación tota) de las dos fases) y transvasar la fase acuosa a un matraz aforado de 250 ml. Lavar la fase etérea con (2 x 5 ml.) de agua, dejando un tiempo de decantación de 15 minutos después de cada lavado (1). Transvasar las soluciones de lavado al matraz aforado, y enrasar con agua destilada. Descartar la fase etérea. Tomar con pipeta 100 ml. y llevar a un embudo de decantación de 250 ml. Adicionar 3 ml. de ácido clorhídrico y extraer tres veces con 25 ml. de éter etílico cada vez, dejando a cada decantación un tiempo de reposo de 15 minutos.
Lavar el conjunto de los extractos etéreos tres veces con 10 ml. de agua destilada (1), dejando un tiempo de decantación de 30 minutos (hasta obtener una separación total de las dos fases) (2). Al final de estos lavados comprobar que el pH de la última fase acuosa separada es aproximadamente 7.
Transvasar la fase etérea a un matraz erlenmeyer de 250 ml. Pasar las aguas de lavado al embudo de decantación y efectuar una extracción con 30 ml. de éter etílico, dejando un tiempo de decantación de 1 hora. Descartar la fase acuosa, lavar la fase etérea dos veces con 25 ml. de agua destilada (1) cada vez, dejando un tiempo de decantación de 30 minutos y comprobando que su pH sea, aproximadamente, 7. Reunir esta fase etérea con la anterior situada en el matraz erlenmeyer.
(1) No emplear mayor cantidad de agua que la indicada. Dependiendo de la naturaleza de los agentes tensio-activos que contenga la formulación pueden presentarse emulsiones o interfases abundantes que dificultan la separación cuantitativa de las fases. Se recurrirá entonces a las técnicas habituales de separación (salado, centrifugado, etcétera.
(2) Caso de formarse una interfase estable, separarla con la fase acuosa, que posteriormente será de nuevo extraída.
Adicionar, a la fase etérea, 60 ml. de tanol y valorar con hidróxido sódico 0,1 N, utilizando azul de bromotimol como indicador. Realizar de forma análoga un ensayo en blanco, empleando 60 ml. de etanol y 40 ml. de agua destilada recientemente hervida.
3.5. Cálculos.

N = normalidad de la solución de NaOH.
a = volumen, en ml., de hidróxido sódico 0,1 N utilizado para la muestra.
b = volumen, en ml., de hidróxido sódico 0,1 N utilizado para el ensayo en blanco, p = peso, en gramos, de la muestra.
d204 = densidad a 20 °C/4 °C.
3.6. Referencias.
1. «Specification for Pesticidas», Who, Geneva, 1961, páginas 334 y 140.
2. «Cipac Handbook», CIPACL, Harpenden, 1970, pág. 255.
3. «Métodos Oficiales de Análisis», Ministerio de Agricultura, Madrid, 1971, pág. 241.
4. DIMETOATO
4.1. Principio.
Purificación por cromatografía en capa gruesa seguida de valoración bromométrica.
4.2. Material y aparatos.
4.2.1. Equipo para cromatografía en capa fina.
4.2.2. Cromatoaplicador con jeringa de 50 µd.
4.3. Reactivos.
4.3.1. Gel de sílice.
4.3.2. Líquido eluyente: n-Hexano/acetona (2,1).
4.3.3. Revelador: Disolución acuosa de Pd Cl2 al 0,2 por 100.
4.3.4. Disolución de ClH al 10 por 100.
4.3.5. Disolución de bromato potásico 0,1 N.
4.3.6. Disolución de tiosulfato sódico 0,1 N.
4.3.7. Disolución acuosa de IK al 50 por 100.
4.3.8. Disolución de almidón.
4.4. Procedimiento.
4.4.1. Mezclar bien en un mezclador 80 g. de gel de sílice y 135 ml. de agua. Aplicar sobre placas una capa de 1 mm. de espesor y secar primero al aire durante 4-5 horas, y finalmente en una estufa de desecación a 120" durante tres horas.
4.4.2. Trazar una línea de 1 mm. de anchura, paralela al borde superior de la placa y situada a 2 cm. del mismo. Eliminar el gel de sílice en una zona de 5 mm. de anchura en los bordes laterales, para evitar una difusión lateral.
Colocar la placa en la cubeta de separación y dejar que suba el líquido hasta la línea marcada. Sacar la placa de la cubeta, dejar evaporar los disolventes, y colocar de nuevo en la cubeta, repitiendo la elución. Sacar de nuevo la placa, evaporar los disolventes, pulverizarla ligeramente con la disolución de Pd Cl2, y secar durante 10 minutos al aire. La primera banda amarilla, a partir de la línea de aplicación, es la zona del dimetoato. Separar cuantitativamente con una espátula la banda cuyo Rf corresponda al dimetoato. ampliada 5 mm. alrededor para asegurarse que se recoge todo el producto.
Llevar el gel de sílice separado a un erlenmeyer de 200 ml. provisto de tapón esmerilado, tratar con 25 ml. de disolución 0,1 N de bromato potásico, 1 g. de KBr y 50 ml. de HCl al 10 por 100, y dejar 10 minutos en la oscuridad a la temperatura ambiente. Añadir 5 ml. de disolución de KI al Sr por 100 y valorar seguidamente con disolución 0,1 N de tiosulfato sódico, usando solución de almidón como indicador. Completar la valoración después de 15 minutos.
Por ello, para eliminar interferencia del color propio de los complejos de paladio, valorar tomando como referencia una disolución patrón preparada de forma análoga.
4.4.3. Recubrir interiormente la cubeta con papel de filtro y añadir 240 ml. de líquido eluyente.
4.4.4. Sobre la línea de partida, situada a 3 cm. del borde de la placa, y que termina a 2 cm. de los bordes laterales, aplicar mediante una pipeta (o mejor mediante un cromatoaplicador) una disolución del problema (aproximadamente 20 mg. de dimetoato) en forma de línea transversal de 16 cm.
4.5. Cálculo.

a = ml. de tiosulfato 0,1 N utilizados para el problema.
b = ml. de tiosulfato 0,1 N utilizados para el blanco.
V = volumen, en ml., de muestra aplicada.
4.6. Referencias.
1. «Zeitschrift für Analytische Chemie», 215, 4.º (1966), página 253.
2. «Journal of Chromatography». 22 (1966), págs. 316-322.
5.ETION
5.1. Principio.
Los compuestos con grupos etoxi unidos al fósforo presentan una banda de absorción intensa entre 1.030 y 1.010 cm-1 para compuestos C2H- —O— P (S). Esta banda de absorción se atribuye a la vibración P —O— (C) en la molécula. El espectro del etion presenta una banda de absorción muy definida, con un máximo a los 1.012 cm-1, que es la que se utiliza para su análisis cuantitativo.
5.2. Material y aparatos.
5.2.1. Espectroíotómetro infrarrojo.
5.2.2. Cubetas de CIÑa o BrK de 0,100 mm. de espesor.
5.2.3. Matraces aforados de 25 ml. con tapón de cristal.
5.3. Reactivos.
5.3.1. Patrón de etion.
5.3.2. Tetracloroetileno para espectrofotometría.
5.4. Procedimiento.
5.4.1. Preparación de la curva patrón:
Transferir cuantitativamente 50, 75, 100, 125 y 150 mg, de patrón de etion a matraces aforados de 25 ml., enrasando posteriormente cada uno con tetracloroetileno para espectrofotometría.
A continuación y por medio de una jeringa, transferir las soluciones patrón a la cubeta, rellenando la cubeta de referencia con tetracloroetileno y obtener el espectrofotograma de cada solución patrón desde 1.050 a 950 cm-1, midiendo la transmitancia a 1.012 cm-1 y a 985 cm-1, tomando esta última como punto base.
Calcular la absorbencia como sigue:

A = absorbancia.
Po = transmitancia a 985 cm-1.
P = transmitancia a 1.012 cm-1.
Obtener la curva patrón llevando absorbancias frente a concentraciones de etion.
5.4.2. Análisis de muestras de etion.
Transferir exactamente de 100 a 150 mg. de etion a un matraz aforado de 25 ml. y proceder como en 5.4.1.
5.5. Cálculos.
Leer la concentración de etion según la curva patrón y calcular el porcentaje de pureza como sigue:

L = lectura en la curva patrón.
P = peso en mg. de la muestra.
Para expresar el resultado en gr/l. multiplicar por 10 y por la densidad a 20 °C/4 °C.
6. HCH POR CROMATOGRAFIA DE GASES
6.1. Principio.
Los isómeros α, β, γ y δ del HCH se separan por cromatografía en fase gaseosa, determinándose cuantitativamente por comparación de sus áreas con el área del pico producido por una cantidad conocida de benzofenona añadida a la disolución problema como patrón interno.
6.2. Material y aparatos.
6.2.1. Cromatógrafo de gases con detector de conductividad térmica.
6.2.2. Microjeringa de 10 µl.
6.2.3. Columna de vidrio según 6.4.4.
6.2.4. Erlenmeyer de 250 ml. con tapón esmerilado.
6.2.5. Agitador magnético.
6.2.6. Embudo de vidrio.
6.2.7. Probeta de 10 ml. con tapón esmerilado.
6.2.8. Matraces aforados de 10 ml.
6.2.0. Pipetas de 10 y 5 ml. de doble enrase.
6.3. Reactivos.
6.3.1. Acetato de etilo para análisis, redestilado.
6.3.2. Benzofenona para análisis.
6.3.3. Patrón de HCH técnico.
6.3.4. Patrones analíticos do los isómeros del HCH.
6.4. Procedimiento.
6.4.1. Preparación de soluciones patrón para la determinación de los factores de respuesta.
En cuatro matraces aforados pesar, aproximadamente, 100 mg. con exactitud de 0,1 mg. de los isómeros del HCH, respectivamente; agregar a cada matraz, aproximadamente, 30 mg. de benzofenona, pesados con exactitud da 0,1 mg., y enrasar a 10 mi, con acetato de etilo.
6.4.2. Determinación de los factores.
Para hallar el factor de respuesta de cada isómero se aplica la siguiente fórmula:

siendo:
Ai = área del pico del isómero correspondiente.
Pb = peso de benzofenona inyectado (µg).
Ab = área del pico de benzofenona.
Pi = peso del isómero inyectado (µg).
6.4.3. Preparación de la disolución problema.
Pesar exactamente M gr. de la muestra, siendo M aproximadamente igual a 25/R (R es la riqueza teórica de la muestra en HCH en tanto por ciento) en un matraz erlenmeyer y agregar 10 ml. de acetato de etilo.
Agitar diez minutos en agitador magnético.
Preparar una disolución de benzofenona en acetato de etilo de M mg/ml. (siendo M aproximadamente igual a 6).
Tomar unos 5 ml. del extracto de la muestra y llevarlos con un volumen exactamente igual de la solución de benzofenona a una probeta de 10 ml. y agitar.
Inyectar en el cromatógrafo I µl.
6.4.4. Condiciones cromatográficas.
Columna: De vidrio de 1 m. de longitud, y aproximadamente de 3 mm. de diámetro interior.
Relleno: S por 100 Apiezón L y 0,5 por 100 Carbowax 20 M Chromosorb W 80/100 mallas.
Gas portador: Helio.
Flujo: 50 ml/min.
Temperatura: Inyector: 250 °C.
Columna: 180 °C.
Detector: 200 °C.
6.4.5. Técnica operatoria:
Una vez estabilizado el aparato, inyectar Iµl. de la disolución problema y proceder a medir el área de los picos.
La tensión en el detector, atenuación del electrómetro, tensiones en mV del registrador y electrómetro y volumen I a inyectar deberán fijarse en cada aparato de forma tal que los picos en el cromatograma entren en escala, siendo el pico máximo de ellos de orden del 65 por 100 de la anchura del registro.
6.5. Cálculo.
Los contenidos del HCH y de cada isómero en la muestra se calculan mediante las expresiones:


Ai = área del pico del isómero.
Fi = factor del isómero.
Ab = área del pico de benzofenona.
i = α, β, γ, δ.
m = concentración, en mg/mg. de benzofenona en acetato de etilo.
M = peso, en gramos, de la muestra.
6.6. Observaciones.
1. Revisar los factores de respuesta al menos una vez al mes.
7.LINDANO POR CROMATOGRAFIA DE GASES
7.1. Principio.
El lindano se analiza por cromatografía en fase gaseosa, determinándose cuantitativamente por comparación de su área con el área del pico producido por una cantidad conocida de benzofenona, añadida a la disolución problema como patrón interno.
7.2. Material y aparatos.
7.2.1. Cromatógrafo de gases con detector de conductividad térmica.
7.2.2. Microjeringa de 10 µl.
7.2.3. Columna de vidrio según 7.4.4.
7.2.4. Erlenmeyer de 250 ml. con tapón esmerilado.
7.2.5. Agitador magnético,
7.2.6. Embudo de vidrio.
7.2.7. Probeta de 10 ml. con tapón esmerilado.
7.2.8. Matraces aforados de 10 ml.
7.2.9. Pipetas de 10 y 5 ml. de doble enrase.
7.3. Reactivos.
7.3.1. Acetato de etilo para análisis, redestilado.
7.3.2. Benzofenona para análisis.
7.3.3. Patrón de lindano.
7.4. Procedimiento.
7.4.1. Preparación de la solución patrón para la determinación del factor de respuesta:
En un matraz aforado pesar aproximadamente 100 mg. de lindano, con exactitud de 0,1 mg.; agregar al matraz aproximadamente 30 mg. de benzofenona, pesados con exactitud de 0,1 mg. y enrasar a 10 ml. con acetato de etilo.
7.4.2. Determinación del factor:
Para hallar el factor de respuesta del lindano se aplica la siguiente fórmula:

Aβ = área del pico de benzofenona.
Pγ = peso del lindano inyectado (µg).
Aγ = área del pico del lindano.
Pβ = peso de benzofenona inyectado (/<g).
7.4.3. Preparación de la disolución problema.
En un matraz erlenmeyer pesar exactamente M g. de la muestra, siendo M aproximadamente igual a 5/R (R es la riqueza teórica de la muestra en lindano, expresada en tanto por ciento), agregar 10 ml. de acetato de etilo. Agitar durante diez minutos en agitador magnético.
Preparar una disolución de benzofenona en acetato de etilo de m mg/ml. (siendo m aproximadamente igual a 30).
Tomar una parte alícuota del extracto de la muestra y llevarlo a una probeta de lo ml. con un volumen de benzofenona correspondiente a una décima parte de esta alícuota, y agitar. Inyectar en el cromatógrafo Iµl.
7.4.4. Condiciones cromatográficas:
Columna: de vidrio, de 1 m. de longitud y aproximadamente de 3 mm. de diámetro interior.
Relleno: 5 por 100 Apiezon L y 0,5 por 100 Carbowax 20 M. en Chromosorb W/100 mallas.
Gas portador: Helio.
Flujo: 50 ml/min.
Temperaturas: Inyector: 250 °C.
Columna: 180 °C.
Detector: 200 °C.
7.4.5. Técnica operatoria:
Una vez estabilizado el aparato, inyectar Iµl. de la disolución problema y proceder a medir el área de los picos.
La tensión en el detector, atenuación del electrómetro, tensiones en mV del registrador y electrómetro, y volumen I a inyectar, deberán fijarse en cada aparato de forma tal que los picos en el cromatograma entren en escala.
7.5. Cálculos.
El contenido de lindano en la muestra se Calcula mediante la expresión:

siendo:
Aγ = área del pico del lindano.
Fγ = factor de respuesta del lindano.
Aβ = área del pico de benzofenona.
M = peso en g. de la muestra.
m = concentración en mg/ml de benzofenona en. acetato de etilo.
7.6. Observaciones.
7.6.1. No deberán aparecer picos de otros isómeros de HCH en el cromatograma.
7.6.2. Revisar el factor de respuesta al menos una vez al mes.
8. DETERMINACION DEL pH EN PRODUCTOS SOLIDOS
8.1. Material y aparatos.
8.1.1. Probeta de 100 ml. con tapón esmerilado.
8.1.2. pH-metro equipado con electrodos de vidrio.
8.2. Procedimiento.
Pesar 1 g. de muestra, transferirla a una probeta conteniendo agua desionizada (50 ml.), completar a 100 ml. y agitar vigorosamente durante un minuto.
Dejar sedimentar y medir el pH del líquido que sobrenada.
8.3. Referencia.
C.I.P.A.C. Edición 1970, pág. 1008.
9. DETERMINACION DEL ZLNEB, MANEB Y PROPINEB EN PRESENCIA DE SALES DE COBRE
9.1. Principio.
Los ditiocarbamatos se valoran por iodometría tras un proceso de descomposición a S2C en medio ácido y en caliente; arrastre de éste y de los gases desprendidos a través de frascos lavadores (dos con acetato de plomo y otro con agua de barita), que retienen los citados gases, y una vez eliminados éstos, se recoge el S2C en un cuarto frasco lavador que contiene potasa etanólica al 12 por 100, formándose etilxantato potásico que se valora, tras neutralización, con iodo 0,1 N.
9.2. Material y aparatos.
9.2.1. Aparato de destilación (figura 9.1).
9.2.2. Matraz de destilación de 500 ml.
9.2.3. Probetas Vigreux con cabezas esmeriladas de 50 ml.
9.2.4. Bomba de vacío o trompa do agua.
9.2.5. Vaso de precipitados de 250 ml.
9.2.6. Buretas contrastadas de 50 ml.
9.2.7. Agitador magnético.
9.2.8. Pipeta de 10 ml.
9.3. Reactivos.
9.3.1. Acido iodhídrico (57 por 100).
9.3.2. Acetato de plomo (10 por 100).
9.3.3. Hidróxido bárico (solución saturada).
9.3.4. Acido acético (30 por 100).
9.3.5. Solución de almidón al 1 por 100 en agua.
9.3.6. Solución etanólica de fenolftaleína.
9.4. Procedimiento.
Montar el aparato representado en la figura 1, los dos primeros frascos lavadores contienen solución de acetato de plomo al 10 por 100, el tercero, solución saturada de agua de barita, y
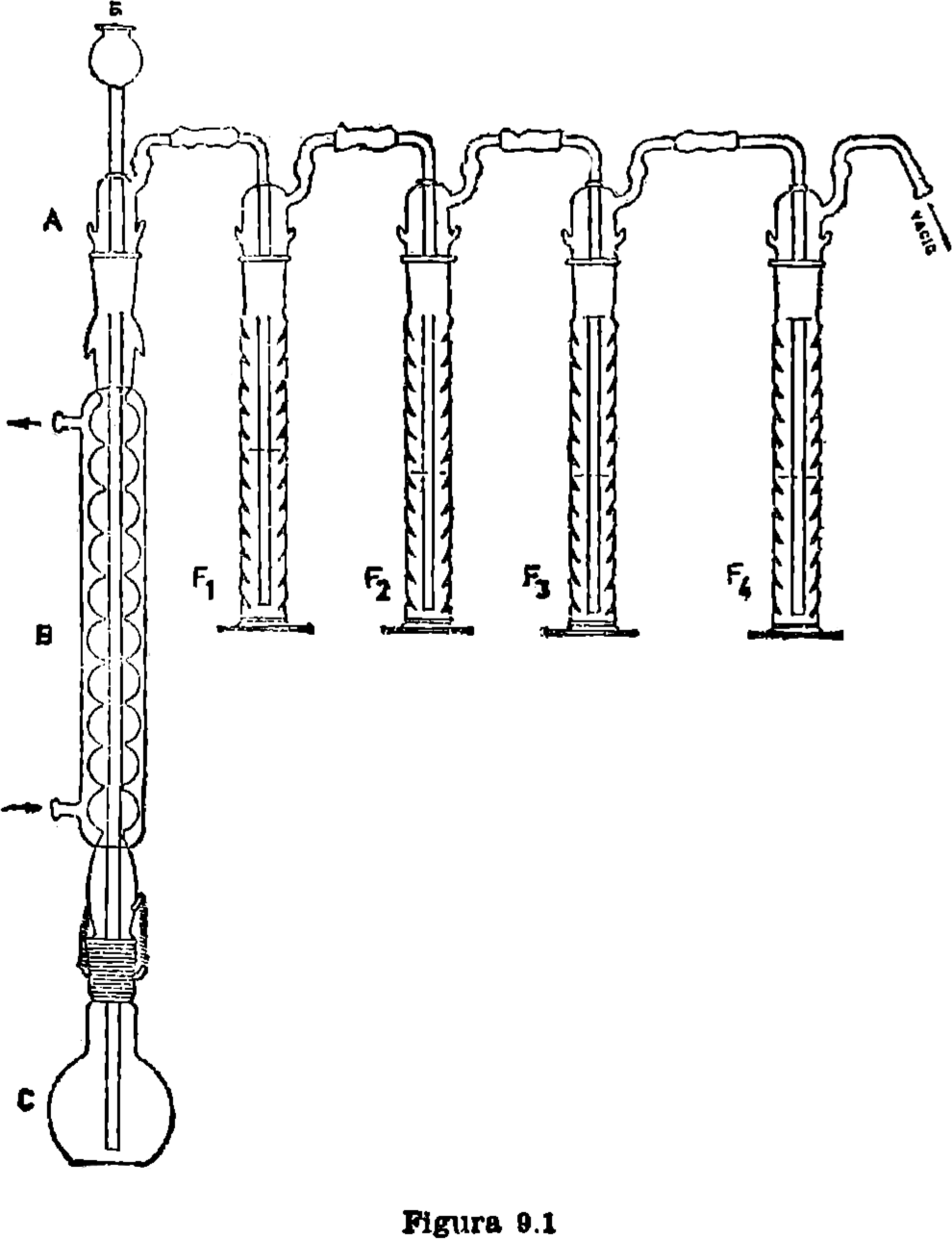
el cuarto, potasa etanólica al 12 por 100 (preparado en el día); en este último se recoge el S2C.
Pesar de 0,2 a 0,3 g. del ditiocarbamato e introducir en el matraz de reacción (c), que debe estar completamente seco, y conectar el vacío (tres burbujas por segundo); a continuación añadir (a) de 30 a 50 ml. de IH al 57 por 100 (d = 1,70) y se empieza a calentar lentamente hasta ebullición, manteniéndola después durante un tiempo de hora y media a dos horas, transcurrido éste, se desconecta el frasco de la potasa etanólica, vertiendo su contenido en un vaso de 500 ml., lavando a continuación el frasco lavador con agua destilada y añadiendo fenolftaleína al mismo, dando por terminado el lavado en el momento en que no exista color azul. Neutralizar con ácido acético al 30 por 100 contenido en una bureta en presencia de fenolftaleína. procurando no acidular la disolución y valorar inmediatamente con una disolución de iodo 0,1 N en presencia de almidón (añadir éste cuando la disolución presente color blanquecino).
El tiempo transcurrido entre el lavado y la valoración no debe ser superior a treinta minutos.
9.5. Cálculos:

K = constante del principio activo.
Zineb: 13,79.
Maneb: 13,27.
Propineb: 14,45.
V = volumen, en ml., de disolución de iodo gastados en la valoración.
V' = volumen, en ml., de disolución de iodo gastados en el ensayo en blanco.
P = peso, en gramos, de la muestra.
N = normalidad exacta de la disolución de iodo.
10. DETERMINACION DEL COBRE (Espectrofotometrla de absorción atómica)
10.1. Principio.
El cobre se extrae de la muestra en columna, como Cu++ y se mide por absorción atómica refiriéndolo a una curva de calibrado. En muestras con cobre orgánico se mineraliza previamente.
10.2. Material y aparatos.
10.2.1. Espectrofotómetro de absorción atómica.
10.2.2. Columna de cromatografía de 1,5 x 30 cm.
10.2.3. Matraz aforado de 1.000 ml.
10.2.4. Pipeta de 10 ml. y doble enrase.
10.2.5. Matraz de 100 ml.
10.3. Reactivos.
10.3.1. Gel de sílice 0,05-0,2 mm. (70-325 mallas).
10.3.2. Algodón hidrófilo.
10.3.3. ClH (1:4) (V/V).
10.4. Procedimiento.
La columna se prepara colocando en el fondo de la misma un trozo de algodón hidrófilo sobre el cual se depositará gel de sílice, hasta una altura de 3 cm. aproximadamente, a continuación se vibra la columna para evitar vías privilegiadas y seguidamente se añade una cantidad de muestra pesada con error de ± 0,0002 g. sobre la gel de sílice (el peso de la muestra será calculado para que contenga aproximadamente unos 50 mg. de Cu). A continuación se eluye con 150 ml. de ClH (1:4), recogiéndose el eluido sobre un matraz aforado de 1.000 ml., enrasándose con agua destilada. Se toman 10 ml. de esta solución y se llevan a un matraz de 100 ml., enrasándose nuevamente. En este segundo matraz se determinará el contenido en cobre, midiendo en espectrofotómetro de absorción atómica a 325,6 nm.
En muestras con cobre en forma orgánica, se mineraliza por calcinación, se recogen las cenizas y se disuelven con ClH (1:4), filtrando a continuación y enrasando en un matraz de 1.000 m3.
Tomar 10 ml. de esta disolución y diluir a 100 ml. siguiendo a continuación el mismo procedimiento que para el cobre inorgánico.
10.5. Cálculos.
La riqueza se calcula de la siguiente manera, en el caso de que se hayan efectuado las diluciones indicadas en el método.

siendo:
R % = riqueza del cobre en tanto por ciento (p/p).
p.p.m. = las partes por millón obtenidas en la curva de calibrado.
p = peso, en gramos, de la muestra.
10.6. Observaciones.
1. En caso de dificultad en el paso del líquido a través de la columna, se puede mezclar la muestra con un coadyuvante de filtración.
11. MANEB EN POLVOS DISPERSABLES
11.1. Principio.
El maneb, disuelto en solución de EDTA tetrasódico, se descompone mediante el acido sulfúrico en ebullición, a etilendiaminsulfato y sulfuro de carbono. Este último se pasa primero a través de un frasco lavador conteniendo sulfato de cadmio para absorber acido sulfhídrico si lo hay, y después a través de un sistema absorbedor conteniendo solución metanólica de hidróxido potásico para formar el metil-xantato potásico, el cual, después de neutralizado con acido acético diluido, se valora con solución standard de yodo.
11.2. Material y aparatos.
11.2.1. Embudo pesador (11.6.2).
11.2.2. Aparato como el Indicado en la figura 11.1.
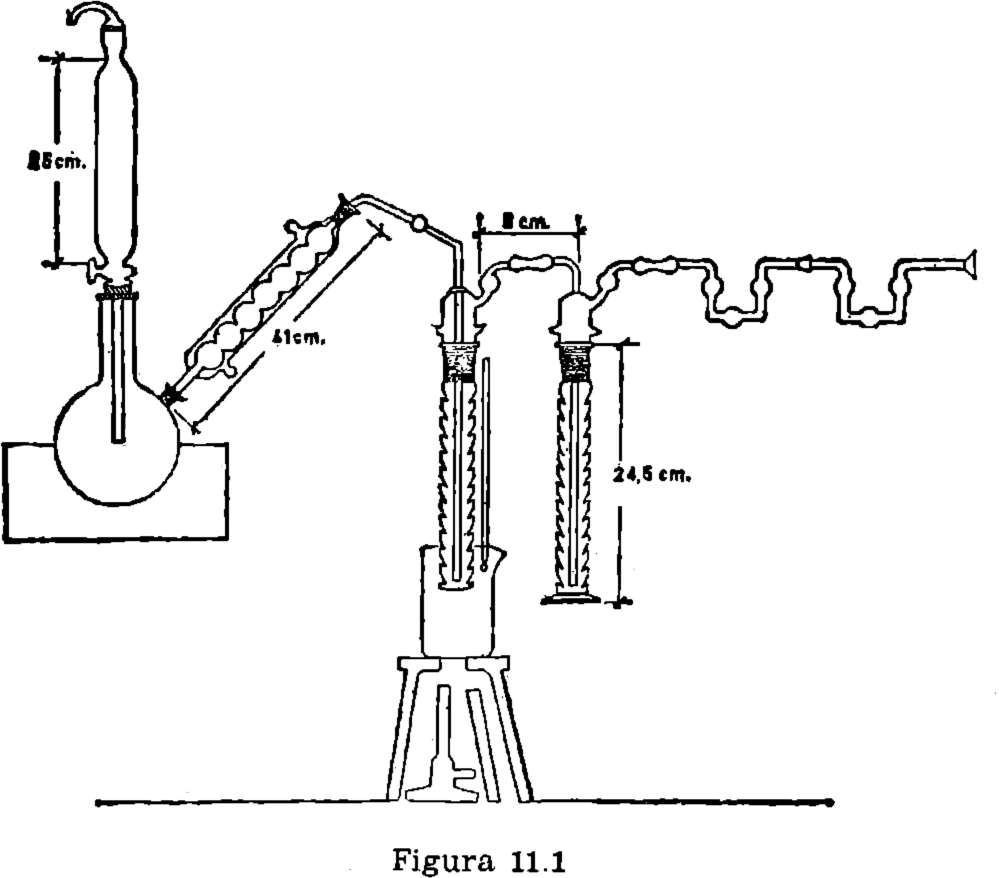
11.2.3. Frasco erlenmeyer de 600 ml.
11.2.4. Dos buretas de 50 ml.
11.3. Reactivos.
11.3.1. Dietilditiocarbamato sódico trihidrato puro (11.8.1).
11.3.2. Acido sulfúrico aproximadamente 4N.
11.3.3. Sal tetrasódica del ácido etilendiamino tetracático EDTA tetrasódico.
11.3.4. Hidróxido potásico. Solución metanólica 2Ni conteniendo menos de 1 p.p.m. de cobre o hierro.
11.3.5. Sulfato de cadmio. Disolver sulfato de cadmio (18,5 g. de 3CdSO4 8H20) en agua destilada (100 ml.).
11.3.6. Acido acético. Solución 10 por 100 p/v.
11.3.7. Fenolftaleína. Solución indicadora al 1 por 100.
11.3.8. Almidón. Solución indicadora.
11.3.9. Yodo. Solución 0,1 N comprobada.
11.4. Procedimiento (11.6.3).
Montar el aparato, como se demuestra en la figura (11.8.4), poniendo sulfato de cadmio (30 ml.) en el primer absorbedor, e hidróxido potásico metanólico (11.6.5) en el segundo (25 ml.) y 5 ml. en cada una de las bolas (11.6.6) Conectar el refrigerante, y calentar el baño de agua, en el que está sumergido el primer absorbedor, hasta 70-80 °C, manteniendo esta temperatura durante la determinación.
Pesar exactamente, mediante el embudo pesador, la cantidad adecuada de muestra que contenga aproximadamente 0,4 g. de maneb (P) y transferirla al matraz de digestión. Montar el embudo decantador con el tubo de entrada de aire (11.6.7) y añadir el EDTA tetrasódico a través del embudo. Dejar un minuto para la dispersión de la muestra, agitando unos instantes en sentido rotatorio el matraz de reacción, para asegurar la dispersión completa en el EDTA; añadir, a través del embudo de decantación, solución de ácido sulfúrico hirviendo (50 ml.) y calentar el matraz inmediatamente. Conectar el tubo final del sistema de absorción a un generador de vacío, controlado de tal manera que pasen a través de los absorbedores tres burbujas de aire por segundo (11.6.8).
Mantener una vigorosa velocidad de reflujo hasta que la muestra se haya descompuesto por entero; se necesitan unos veinticinco minutos. Cerrar el agua del refrigerante durante dos o tres minutos, llenando de este modo con vapor el refrigerante y el absorbedor de sulfato de cadmio para asegurar el transporte de todo el sulfuro de carbono.
Quitar el mechero, desconectar el sistema de absorción y, sin demora, proceder a la determinación del sulfuro de carbono como sigue:
Lavar cuidadosamente el contenido del absorbedor de potasa metanólica y las bolas pasándolas a un erlenmeyer de 600 ml. usando agua destilada (300-400 ml.); añadir una o dos gotas de fenolftaleína, neutralizar con ácido acético mediante una bureta, y añadir tres gotas en exceso.
Agitando continuamente, valorar la solución inmediatamente, por ejemplo, en el tiempo de dos minutos (11.6.9), y con la solución de yodo. Cerca del punto final, añadir solución indicadora de almidón y completar la valoración (en V ml.).
Proceder a una determinación en blanco omitiendo la muestra (V' ml.).
11.5. Cálculo.

N = normalidad de la solución de yodo.
V = volumen en ml. de solución de iodo gastados en la valoración.
V' = volumen en ml. de solución de iodo gastados en la prueba en blanco.
11.6. Observaciones.
11.6.1. Comprobar la pureza de dietilditiocarbamato de sodio como sigue: Disolver unos 0,5 g. de material en agua (100 mililitros) y valorar directamente con solución acuosa 0,1 N de yodo usando almidón como indicador, 1 ml. solución 0,1 N de yodo <> 0.02253 g. de dietilditiocarbamato sódico trihidrato.

P1 = peso en g. de dietilditiocarbamato.
V1 = volumen en ml. de solución de yodo gastados en la valoración.
11.6.2. El modelo preferido consiste en una «navecilla de pesada» que tiene una salida en un extremo a través del cual la muestra se pasa al matraz.
11.6.3. Verificar todo el procedimiento usando dietilditiocarbamato sódico; si se lleva a cabo correctamente se obtendrá una recuperación de 99 a 101 por 100.
11.6.4. Todas las juntas deben ser impermeables al gas.
Se puede usar ácido fosfórico para este propósito. También es efectivo untar con vaselina o silicona, si se usan en pequeñas cantidades.
11.6.5. El principal absorbedor metanólico deberá enfriarse sumergiéndolo en un recipiente con agua y hielo.
11.6.6. Se puede usar cualquier sistema eficiente de absorción. El absorbedor de potasa metanólica y las «bolas» deben estar secos, o se deben lavar con metanol antes del uso.
11.6.7. El tubo de entrada de aire llegará tan cerca como sea posible del fondo del matraz.
11.6.8. Alternativamente el tubo «de entrada» del embudo decantador se puede conectar a una fuente controlada de aire comprimido.
11.6.9. Si la valoración no se completa en el tiempo de dos minutos desde la adición del ácido acético, la solución se debe rechazar y repetir el análisis.
11.7. Referencia.
1. CIPAC 61/3/MI, edic. 1870.
12. DETERMINACION DE CLORO ORGANICO TOTAL
12.1. Principio.
Transformación del cloro contenido en la muestra en cloruro sódico, según el procedimiento de Stephanow, por ebullición con alcohol isopropílico y sodio, y valoración volumétrica.
Para aplicación a la determinación cuantitativa indirecta de los plaguicidas clorados, para los que no se dispone de métodos específicos.
12.2. Material y aparatos.
12.2.1. Tres matraces erlenmeyer de 100, 200 y 500 ml.
12.2.2. Agitador eléctrico poco revolucionado.
12.2.3. Baño de agua eléctrico.
12.2.4. Matraz de fondo redondo, de 500 ml. con boca esmerilada.
12.2.5. Refrigerante recto, con tubo interior ancho, de 250 milímetros, esmerilado.
12.2.6. Embudo de llave de 50 ml.
12.2.7. Pipetas de dos enrases de 10, 20 y 100 ml.
12.2.8. Probetas de 25 y 5 ml.
12.2.9. Bureta de 50 ml.
12.3. Reactivos.
12.3.1. Benceno para análisis exento de cloro y de tiofeno.
12.3.2. Isopropanol para análisis.
12.3.3. Isopropanol al 50 por 100 v/v en agua destilada.
12.3.4. Sodio metálico para análisis.
12.3.5. Solución alcohólica de fenolftaleína al 1 por 100.
12.3.6. Acido nítrico diluido: 100 ml. de ácido nítrico concentrado (d = 1,38) más 100 ml. de agua destilada.
12.3.7. Solución de nitrato de plata 0,1N.
12.3.8. Solución de sulfocianuro potásico 0,1N.
12.3.9. Solución de sulfato férrico al 10 por 100.
12.4. Procedimiento.
Para analizar productos sólidos, pesar con precisión de 0,1 miligramos una cantidad de muestra que contenga aproximadamente 0,36 g. de cloro (siendo R % la riqueza supuesta en compuesto de peso molecular Pm con n átomos de cloro, tomar unos Pm/nR, g. de muestra) en un tubo de ensayo, pesar, vaciarlo en un erlenmeyer de 200 ml. y destarar el tubo vacío. Añadir, con pipeta de dos enrases, 100 ml. exactos de benceno, mantener agitando una hora y filtrar rápidamente por papel, tapando el embudo con un vidrio de reloj. No lavar el filtro. Pasar 10 ml. exactos del líquido filtrado al matraz de 500 ml., evaporar en el baño de agua la mayor parte del benceno, sin llegar a desecar y sacarlo del baño.
Añadir 25 ml. de isopropanol puro, agitar, agregar 2,5 g. de sodio cortado en tiras finas y conectar el refrigerante a reflujo. Montar el aparato sobre el baño y mantenerlo en ebullición suave una hora, con agitación frecuente y agregar a través del refrigerante, 10 ml. de isopropanol al SO por 100 a razón de 1-2 gotas por segundo. Retirar tíél baño, lavar el refrigerante con agua destilada y desconectarlo. Calentar otros treinta minutos el matraz abierto, comprobar que no huele a isopropanol, enfriar, agregar dos gotas de solución de fenolftaleína, neutralizar con ácido nitrico, añadir.unos lOYnl, de exceso y, después de agitar, 20 ml. exactos de solución 0,1N de nitrato de plata.
Poner el matraz en el baño de agua a 80-90° y mantener veinte minutos, agitando varias veces para coagular el precipitado de cloruro de plata formado. Enfriar, filtrar por papel a un erlenmeyer de 500 ml. lavando varias veces con agua destilada. el matraz y el filtro, añadir al líquido 5 ml. de solución de sulfato férrico y valorar, hasta coloración rojiza débil persistente, con el volumen necesario de solución 0-1N de sulfocianuro potásico.
12.5. Cálculos.
Calcular el contenido en cloro de la muestra, o el contenido en componente clorado, expresados ambos en porcentaje.


N = normalidad exacta de la solución de nitrato de plata.
N' = normalidad exacta del sulfocianuro potásico.
V = volumen en ml. de solución de sulfocianuro potásico.
P = peso en g. de la muestra.
Pm = peso molecular del componente clorado.
n = número de átomos de cloro en la molécula del componente clorado.
12.6. Observaciones.
Este método deberá completarse con una determinación cualitativa que identifique el compuesto (cromatografía en capa fina o en papel; extracción y determinación de constantes físicas).
Hay que considerar la posibilidad de otros componentes halogenados en la formulación.
Si el componente activo se agregó como producto técnico, con isómeros u otros compuestos halogenados, dará resultados altos.
Si la formulación es liquida,.y miscible con isopropanol, diluir la muestra en un matraz aforado de 100 ml., con isopropanol puro, enrasar, agitar y tomar 10 ml. con los que se continúa el análisis.
Si la muestra contiene azufre elemental o en la molécula de algún componente, oxidar dos veces con 5 ml. de agua oxigenada al 30 por 100, el liquido diluido de tratar con sodio, hervir quince minutos después de cada adición, y continuar la valoración en la forma indicada.
Si el producto halogenado es poco soluble en benceno, se hará la extracción con otro disolvente volátil adecuado.
Si existe bromo, también se valora cuantitativamente.
12.7. Referencias.
1. O.M.S., «Specifications for Pesticides», M/16, 3.ª edición (1967), 288-290.
2. A. O. A. C., «Official Methods oí Analysis», 10.a ed. (1965), 4.187-89.
3. Ruiz Castro, «A. Bol. Pat. Veg. y Ent. Agrie.», 27-205-392 (1964).
13. DETERMINACION DE FOSFORO TOTAL (Provisional)
13.1. Principio.
Mineralización de los componentes orgánicos de la muestra por ebullición con ácido sulfúrico y nitrato sódico y valoración colorimétrica del azul de molibdeno correspondiente al ácido fosfórico obtenido.
Para aplicación a la determinación cuantitativa indirecta de los plaguicidas, para los que no se dispone de métodos específicos.
13.2. Material y aparatos.
13.2.1. Matraz Kjeldahl de 300 ml.
13.2.2. Probeta de 25 ml.
13.2.3. Matraces aforados de 50 y 50 ml.
13.2.4. Pipetas de dos enrases de 1 y 2 ml.
13.2.5. Pipetas de Mohr de 10 ml.
13.2.6. Espectrofotómetro o colorímetro (filtros 625 a 675 nm).
13.2.7. Cubetas de 10 mm. de espesor.
13.3. Reactivos.
13.2.1. Acido sulfúrico concentrado, reactivo análisis.
13.3.2. Nitrato sódico, reactivo análisis.
13.3.3. Solución patrón de fosfato monopotásico: 0.4394 g. de fosfato monopotásico puro y seco, disuelto hasta 1 litro en matraz aforado con agua destilada (1 ml. contiene 0,1 mg. de fósforo).
13.3.4. Solución de molibdato amónico al 5 por 100: 25 g. de molibdato amónico R. A. disueltos en 300 ml. de agua destilada, a los que se agrega una solución fría de 75 ml. de ácido sulfúrico concentrado diluidos a 200 ml. sobre agua destilada.
13.3.5. Solución de hidroquinona al 0,5 por 100: 2,5 g. de hidroquinona pura disuelta en 500 ml. de agua destilada y con adición de cinco gotas de ácido sulfúrico concentrado. No usar cuando está muy coloreado.
13.3.6. Solución de sulfito sódico al 20 por 100: 100 g. de sulfito sódico disueltos en unos 400 ml. de agua destilada, completando a 500 y filtrándolo. Mantener el frasco bien cerrado.
13.4. Procedimiento.
13.4:1. Aplicable a las formulaciones que contienen una cantidad de fósforo igual o mayor al 2 por 100.
Emplear una cantidad de muestra que contenga aproximadamente 0,1 g. de P. (Para analizar una formulación de riqueza supuesta R % en sustancia activa de peso molecular Pm con n átomos de fósforo, tomar aproximadamente 0,33 Pm/nR.)
La muestra se pesa con precisión de 0,1 mg., empleando un pequeño vasito de vidrio (unos 20 x 20 mm.) que se introduce en el matraz, Kjedahl, procurando no manchar el cuello, agregar 25 ml. de ácido sulfúrico concentrado y calentar en vitrina a fuego directo con llama de poco aire, al principio suavemente hasta que no se forme espuma, y luego, a ebullición lenta hasta formación de humos blancos durante una hora.
Dejar enfriar unos 5 minutos, agregar aproximadamente un gramo de nitrato sódico, procurando que caiga sobre el líquido, y calentar a ebullición suave hasta formación de humos blancos densos durante varios minutos. Repetir esta adición hasta conseguir que el líquido quede amarillo muy claro en caliente e incoloro en frío.
Si no se consigue con cuatro adiciones de 1 g. de nitrato, puede continuarse agregando porciones de aproximadamente 1 ml. de ácido nítrico concentrado, dejando enfriar unos 5 minutos antes de cada adición. Dejar enfriar completamente el líquido y agregar con cuidado pequeñas porciones de agua destilada, agitando cada vez, hasta diluir a unos 100-150 ml. Añadir unas perlas de vidrio y hervir 5 minutos.
Pasar todo el líquido frio a un matraz aforado de 500 ml., así como tres lavados del matraz Kjeldahl con porciones de unos 50 ml. de agua destilada, completar el volumen y mezclar bien Esta solución deberá ser incolora y transparente, salvo los casos en que se analiza una formulación en polvo cuyos diluyentes minerales producirán una suspensión, que debe dejarse sedimentar, centrifugar o filtrar una porción, para que el liquide quede perfectamente transparente.
Preparar tres matraces aforados de 50 ml., en uno de los cuales se añaden solamente los reactivos para que sirva de blanco al medir el color, en otro se ponen 2 ml. exactos de la solución patrón de fosfato monopotásico y en el tercero 1 ml. exacto de la solución incolora y transparente de la muestra.
Agregar a cada matraz, sucesivamente y por este orden, 2.5 ml. de solución de molibdato amónico. 2,5 ml. de solución de hidroquinona y 2,5 ml. de solución de bisulfito sódico, medidos con pipetas Mohr, mezclando bien después de cada adición y de enrasar con agua destilada.
Dejar reposar unos 25 minutos, pasar los líquidos a cubetas marcadas de 10 mm. de espesor y medir a los 30 minutos las absorbencias del patrón y de la muestra respecto al blanco con un espectrofotómetro a 650 nm. o con un colorímetro con filtro de transmisión máxima a 325-675 nm.
13.4.2. Aplicable a las formulaciones que contienen una cantidad de fósforo menor del 2 por 100.
Se emplea una cantidad de muestra que contenga aproximadamente 0,01 g. de fósforo, que se trata en la misma forma que en el caso anterior, pero empleando para la formación del color 10 ml. exactos, de los 500 a que se diluyó la solución de la muestra.
Para evitar que el ácido sulfúrico que contienen estos 10 mililitros interfiera la formación del color, se valoran otros 10 mililitros con solución de sosa 4 N, empleando como indicador rojo de metilo y se agrega a los 10 ml. que se van a valorar, antes de añadir los reactivos, el volumen de sosa hallado, menos 0,5 ml.
13.5. Cálculos.
13.5.1. Para formulaciones que contienen una cantidad de fórforo igual o mayor al 2 por 100. Calcular el contenido en fósforo de la muestra o el contenido en componentes fosforados, expresados ambos en porcentaje.


P = peso en g. de la muestra.
Am = absorbancia de la muestra.
Ap = absorbancia del patrón.
Pm = peso molecular del componente fosforado.
n = número de átomos de fósforo en la molécula del componente fosforado.
13.5.2. Para formulaciones que contienen una cantidad de fósforo menor del 2 por 100, dividir los resultados de las fórmulas anteriores por 10.
13.6. Observaciones.
Este método deberá completarse con una determinación cualitativa que identifique el compuesto (cromatografía en capa fina o en papel; extracción y determinación de constantes físicas).
Hay que considerar la posibilidad de otros componentes fosforados en la formulación.
Si el componente activo se agregó como producto técnico con isómeros u otros compuestos fosforados, dará resultados altos.
13.7. Referencias.
1. A. O. A. C., «Official Methods of Analysis-, 10.ª ed. (1965), 6.060-64.
2. Gunther, F. A., y Blinn, R. C., «Analysis of Insecticides and Acaricides» (1965), 538-540.
14. VALORACIÓN DE COBRE
14.1. Principio.
Valoración del iodo liberado por la acción del ion cúprico sobre un ioduro, en medio acético.
Para la valoración del contenido en cobre en las formulaciones normales, que contengan «azufres negros».
14.2. Material y aparatos.
14.2.1. Matraz erlenmeyer de 200 ml., con agitador.
14.2.2. Matraz erlenmeyer de 500 ml.
14.2.3. Matraz aforado de 250 ml.
14.2.4. Pipeta de dos enrases de 25 ml.
14.2.5. Vaso de 200 ml.
14.2.6. Cuatro pipetas de Mohr de 10 ml.
14.2.7. Bureta de 50 ml.
14.3. Reactivos.
14.3.1. Acido clorhídrico diluido; 200 ml. de ácido clorhídrico concentrado, para análisis, diluidos a 1 litro.
14.3.2. Disolución saturada de carbonato sódico-. 250 g. de carbonato sódico cristalizado, para análisis; en frasco de 1 litro con tapón de politeno, añadir 800 ml. de agua destilada o desionizada, agitar unos minutos y dejar sedimentar el exceso de sal.
14.3.3. Disolución de carbonato sódico cristalizado, para análisis, al 10 per 100.
14.3.4. Acido acético glacial para análisis.
14.3.5. Disolución de ioduro potásico para análisis al 30 por 100.
14.3.6. Indicador de almidón; desleír bien 1 g. de almidón soluble en unos 10 ml. de agua destilada y añadir agitando sobre otros 100 ml. de agua destilada hirviendo, continuando la ebullición 2 minutos, Enfriar y conservar bajo una capa de tolueno; tomar con pipeta.
14.3.7. Solución de tiosulfato sódico 0,1 N. Disolver 24,32 g. de sal pura cristalizada en matraz aforado de 1 litro con unos 900 ml. de agua destilada, completar el volumen y mezclar. Esperar unos 10 días antes de determinar su factor, frente a un patrón primario puro, por las técnicas usuales, lo que debe repetirse con frecuencia.
14.4. Procedimiento.
14.4.1. Compuestos cúpricos insolubles en agua (carbonato, oxicloruro, sulfatos básicos).
Pesar con precisión de 0,1 mg. una muestra que contenga aproximadamente 1 g de Cu, agitar una hora en el erlenmeyer de 200 ml. con 50 ml. de ácido clorhídrico diluido; si la cantidad de muestra no permite agitar bien, pueden emplearse 100 ml. Filtrar por papel y lavar con agua destilada el matraz y el filtro hasta que el líquido filtrado no dé reacción de cobre. Diluir a unos 200 ml., hervir 10 minutos, enfriar y pasar a un matraz aforado de 250 ml., así como tres porciones de agua destilada de unos 10 ml. con que se lava el recipiente en que se hirvió, completar el volumen y mezclar bien. Tomar 25 ml. exactos en un vaso y diluir a doble volumen con agua destilada, añadiendo a continuación, agitando, solución saturada de carbonato sódico hasta que desprenda poco anhídrido carbónico, y luego, gota a gota, solución al 10 por 100 hasta que se observe ligero precipitado que no desaparece al agitar, disolverlo con unas gotas de ácido acético glacial y añadir un exceso de 2 mililitros. Añadir 8 a 10 ml. de disolución de ioduro potásico al 30 por 100 y agitar 2 minutos, con lo que se forma precipitado amarillo y coloración parda en el líquido.
Valorar el lodo liberado con el volumen necesario de disolución de tiosulfato 0,1 N cuya normalidad exacta haya sido determinada recientemente. Cuando la tonalidad parda es poco perceptible, añadir 2 ml. de indicador almidón, continuando la valoración hasta la desaparición del color azulado durante 2 minutos.
14.4.2. Compuestos cúpricos insolubles acompañados de arsenicales.
Añadir a los 25 ml. de disolución a valorar, 5 ml. de agua oxigenada al 30 por 100 y hervir, añadir 100 ml. de agua destilada y continuar la ebullición hasta reducir el volumen a la mitad. Una vez frío, continuar la valoración en la forma indicada.
14.4.3. Sulfato cúprico.
Pesar exactamente una cantidad de muestra que contenga aproximadamente 4 g. de SO4Cu.5H2 disolverlo por agitación durante unos minutos, con 150 ml. de agua destilada, pasar a un matraz aforado de 250 ml. (filtrando y lavando el filtro si la solución no es transparente) y completar el volumen con agua destilada. Tomar 25 ml., con pipeta de dos enrases, que se hierven en un vaso hasta reducir su volumen a unos 10 ml., que se neutraliza con carbonato sódico, continuando la valoración en la forma descrita.
14.5. Cálculo.
Calcular el contenido en cobre, expresado en porcentaje o como SO4Cu.5H2O.


V = volumen en ml. de disolución de tiosulfato.
N = normalidad exacta de la disolución de tiosulfato.
P = peso en g. de la muestra.
14.6. Observaciones.
Si la muestra contiene carbonatos, añadir previamente y con cuidado la cantidad de ácido clorhídrico diluido necesario para que termine la efervescencia, y después los 50 ml. que se indican.
14.7. Referencias.
1. F.A.O., «Boletín Fitosanitario» (16), 11-11 (1963).
2. A.O.A.C., «Official Methods of Analysis», 11.ª ed., 6.015 y 6.056 a 6.086 (1070).
3. Ruiz Castro, A., «Bl. Pat. Veg. y Ent. Agric.», 24, 219-250 (1959).
15. DETERMINACION DE MERCURIO EN DESINFECTANTES
15.1. Principio.
Disolución del mercurio contenido en la muestra por ebullición con ácido sulfúrico concentrado y agua oxigenada al 30 por 100 y precipitación en forma de sulfuro.
Aplicable a los desinfectantes mercuriales de semillas.
15.2. Material y aparatos.
15.2.1. Matraz erlenmeyer de 200 ml. con refrigerante de aire (tubo de unos 12 mm. de diámetro y 600 mm. de longitud) Con unión esmerilada.
15.2.2. Vaso de precipitados, en vidrio fino, de 500 ml.
15.2.3. Aparato de Kipp con frasco lavador.
15.2.4. Crisol de vidrio filtrante con placa de porosidad 4.
15.3. Reactivos.
15.3.1. Acido sulfúrico concentrado para análisis.
15.3.2. Agua oxigenada al 30 por 100.
15.3.3. Solución de permanganato potásico al 3 por 100.
15.3.4. Sulfuro ferroso.
15.3.5. Acido clorhídrico diluido: Diluir 200 ml. de ácido clorhídrico concentrado para análisis, hasta 1 litro.
15.3.6. Alcohol etílico de 96 por 100.
15.3.7. Sulfuro de carbono para análisis.
15.4. Procedimiento.
Pesar, con precisión de 0,1 mg„ unos 2 g. de muestra en el matraz erlenmeyer, añadir 10 ml. de ácido sulfúrico, adaptar el refrigerante y girar el matraz para mojar bien la muestra. Añadir gota a gota, a través del refrigerante, 5 ml. de agua oxigenada y girar el matraz para mezclar. Cuando la reacción se suaviza, calentar en llama pequeña 20 minutos, añadir otros 5 ml. de agua oxigenada y continuar la calefacción hasta que se ha destruido toda la materia orgánica (solución blanca), añadiendo para ello más agua oxigenada si es necesario.
Dejar enfriar, lavar el refrigerante con unos 30 ml. de agua destilada y pasar el líquido al vaso, así como tres porciones de unos 10 ml. de agua destilada con que se lava el matraz. Si no fuese el líquido transparente, se pasará filtrando por papel, lavando bien el filtro.
Diluir a unos 200 ml. con agua destilada y destruir el exceso de agua oxigenada, añadiendo, gota a gota, solución de permanganato potásico hasta color rosado débil persistente. Precipitar con corriente de ácido sulfhídrico, lavado con agua destilada, durante 20 minutos, y filtrar por el crisol de vidrio, previamente tarado después de secarlo 30 minutos a 105-110°, lavar con agua destilada, tres veces con alcohol de 96 por 100 y cuatro veces con sulfuro de carbono, sin hacer succión, o muy ligera, con este último disolvente.
Secar 30 minutos a 105-110°, enfriar en desecador y pesar el sulfuro de mercurio obtenido.
15.5. Cálculos.
Calcular el contenido en mercurio de la muestra, o el contenido en componente activo, expresados ambos en porcentaje.


P = peso en g. de la muestra.
P' = peso en g. del sulfuro de mercurio obtenido.
Pm = peso molecular del componente activo.
n = número de átomos de mercurio en la molécula del componente activo.
15.6. Observaciones.
Si los resultados son inferiores a los normales, deberá repetirse la valoración con el residuo de la primera filtración, seco.
15.7. Referencias.
1. A.O.A.C., «Official Methods of Analysis», 11.ª ed. (1970), 6.173.
2. A.O.A.C., «Official Methods of Analysis», 11.ª ed. (1970), 36.327.
3. F.A.O., «Boletín Fitosanitario» (1), 9-19 (1961).
10. VALORACION DE SEVIN (CARBARILO) (Provisional)
16.1. Principio.
Transformación en metilamina, por calefacción en medio alcalino, que arrastrada por una corriente de nitrógeno, se recoge sobre solución de ácido bórico y se valora con ácido clorhídrico.
Para aplicación a la valoración de Sevin en el producto técnico y en sus formulaciones normales.
16.2. Material y aparatos.
16.2.1. Botella de nitrógeno puro, con un manorreductor normal y otro especial que permita trabajar con toda seguridad a presiones inferiores a 0,1 kg/cm2.
16.2.2. Matraz fondo redondo de 125 ml. con dos bocas esmeriladas, una pequeña por la que penetra hasta cerca del fondo el tubo para entrada del nitrógeno, y otra grande para su unión a la columna.
16.2.3. Manta de calefacción eléctrica de 200 W., con reóstato o transformador para su regulación, para el matraz de 125 ml.
16.2.4. Columna con dientes Vigreaux, de unos 20 mm. de diámetro y 300 mm. de longitud, aislada con cinta de amianto, con unión esmerilada al matraz y salida horizontal, en su extremo superior, de tubo de vidrio de unos 8 mm. y unos 200 milímetros de longitud, curvada verticalmente hacia abajo al final, que se acopla con un pequeño esmerilado esférico al tubo de desprendimiento, terminado en dedal de vidrio fritado de poro grueso, que se sumerge en la probeta.
16.2.5. Probeta de 250 a 300 mi, con un enrase a 150 ml.
16.2.6. Bureta de 50 ml.
10.3. Reactivos.
10.3.1. Solución de hidróxido potásico aproximadamente N en dietilenglicol: disolver 66 g. de hidróxido potásico en lentejas para análisis, en 40 ml. de agua destilada y completar el volumen a 1 litro con dietilenglicol puro.
16.3.2. Solución de ácido bórico al 2 por 100: disolver 20 g. de ácido bórico para análisis, en unos 950 ml. de agua destilada caliente, enfriar, añadir 10 ml. de indicador verde bromocresol (solución alcohólica al 0,1 por 100), llevar a color verde esmeralda por adición de ácido clorhídrico 0,1 N y completar a 1 litro con agua destilada.
16.3.3. Solución de ácido clorhídrico 0,1 N: diluir 8,4 ml. de ácido concentrado para análisis, en agua destilada hasta completar 1 litro y determinar su factor de normalidad.
16.4. Procedimiento.
Pesar con precisión de 0,1 mg. una cantidad de muestra que contenga aproximadamente 0,5 g. de producto activo, y añadir 50 ml. de la solución de hidróxido potásico, así como algunas cuentas de vidrio y remover para homogeneizar. Poner el matraz en la manta calefactora y conectar a la columna y tubo de desprendimiento, poniendo grasa de silicona en los esmeriles. Poner en la probeta unos 150 ml. de solución de ácido bórico e iniciar la calefacción y el paso de nitrógeno a 50-80 ml. por minuto.
Se mantiene en ebullición 30 minutos y se desconecta el tubo de desprendimiento antes de cortar el paso de nitrógeno, recogiendo el liquido de la probeta y los de lavar tres veces el tubo y la probeta con más solución de ácido bórico, en un matraz erlenmeyer de 500 ml., donde se valora con ácido clorhídrico 0,1 N, hasta conseguir que tenga exactamente el color inicial de la solución de ácido bórico.
16.5. Cálculo.
Calcular el contenido en Sevin expresado en porcentaje.

P = peso en g. de la muestra.
V = volumen en ml. de ácido clorhídrico 0,1 N.
N = normalidad exacta del ácido clorhídrico 0,1 N.
16.6. Referencias.
1. Euiz Castro, A., y Rodríguez Matia, E., Bol. Pat. Veg. y Ent. Agric.», 28-203-222 (1965).
17. VALORACION DE METALDEHIDO (Provisional)
17.1. Principio.
Despolimerización, por calefacción en medio ácido, a acetaldohído que se recoge en solución de bisulfito sódico, cuyo exceso se oxida, y valoración del combinado con el acetaldehído, por iodometría.
Para aplicación a la valoración del metaldehído en las formulaciones helícidas.
17.2. Material y aparatos.
17.2.1. Matraz de destilación de 250 ml. con tubo de salida curvado hacia abajo verticalmente.
17.2.2. Tubo de absorción de bolas, según Meyer.
17.2.3. Baño de agua.
17.2.4. Trompa de agua.
17.2.5. Matraz aforado de 100 ml.
17.2.6. Pipeta de dos enrases de 20 ml.
17.2.7. Erlenmeyer de 100-150 ml.
17.3. Reactivos.
17.3.1. Acido fosfórico, para análisis (d = 1,5 ó 1,7).
17.3.2. Solución concentrada de bisulfito sódico, para análisis, al 40 por 100.
17.3.3. Bicarbonato sódico, para análisis.
17.3.4. Indicador de almidón: desleir bien 1 g. de almidón soluble en unos 10 ml. de agua destilada y añadir agitando sobre otros 100 ml. de agua destilada hirviendo, continuando la ebullición dos minutos Enfriar y conservar bajo una capa de tolueno; tomar con pipeta.
17.3.5. Solución de iodo 0,1 N.
17.4. Procedimiento.
Pesar, al 0,1 mg., una cantidad de muestra pulverizada que contenga aproximadamente 0,1 g. de metaldehído, empleando para ello un tubo de ensayo pequeño, que se vacía en el matraz, sin manchar el cuello de éste, y se destara. Agregar 100 ml. de agua destilada y 5 ml. de ácido fosfórico d = 1,5 (ó 4 ml. d = 1.7) y agitar para homogeneizar. Tapar la boca del matraz con un tapón atravesado por un tubo de vidrio, estirado en punta fina, que llegue cerca del fondo y unir el extremo (doblado hacia abajo) del tubo de salida del matraz con el tubo de absorción, que contiene 20 ml. de agua destilada mezclados con 2 ml. de solución de bisulfito sódico al 40 por 100, y que a su vez está unido a la trompa de vacío regulada para obtener una corriente de aire estable cuyas burbujas puedan contarse.
Mantener el matraz en el baño de agua en ebullición y con arrastre de aire, durante tres horas, pasando a continuación el líquido del tubo de absorción a un matraz aforado de 100 ml., así como los líquidos de lavarle tres veces con agua destilada. Completar el volumen, mezclar bien y pasar 20 ml. exactos a un matraz erlenmeyer de 100-150 ml., añadiéndole aproximadamente 1 ml. de indicador de almidón y la cantidad de solución de iodo 0,1 N para conseguir coloración ligeramente azulada. No se anota el reactivo consumido, pero deberá observarse que es superior a 3 ml.
Añadir 1 g. de bicarbonato sódico y valorar el bisulfito liberado con el volumen necesario de solución 0,1 N de iodo para obtener coloración azul persistente.
17.5. Cálculo.
Calcular el contenido en metaldehido, expresado en porcentaje.

P = peso en g. de la muestra.
V = volumen en ml. de solución 0,1 N de iodo.
N = normalidad exacta de la solución 0,1 N de iodo.
17.0. Referencias.
1. Rodríguez Matia, E., •Bol. Pat. Veg, y Ent. Agric.», 29435-442 (1966).
Métodos físicos
1. ESTABILIDAD DE UNA EMULSION
1.1. Principio.
Obtención de la emulsión en condiciones definidas y observación de su estabilidad a temperatura constante.
Para aplicación a las formulaciones de plaguicidas emulsionables en agua.
1.2. Material y aparatos.
1.2.1. Vaso de precipitados, de vidrio grueso, de 250 ml. y 80-65 mm. de diámetro interior con una marca de aforo a 100 mililitros.
1.2.2. Pipeta de Mohr de 5 ml.
1.2.3. Probeta de 100 ml.
1.2.4. Embudo de separación de 100 ml.
1.2.5. Estufa (o baño) graduable a 30° ± 1°.
1.3. Reactivos.
1.3.1. Agua dura patrón. Disolver 0,304 g. ClCa (anhidro) y 0,139 g. Cl3Mg.6H2O en agua destilada o desionizada, y completar hasta 1 litro. Deberá mantenerse varios horas en la estufa a 30° antes de su empleo.
1.4. Procedimiento.
1.4.1. Aplicable a los productos emulsionables que se han de agregar sobre el agua en proporción igual o superior al 5 por 100.
En el vaso de 250 ml. conteniendo unos 80 ml. de agua dura patrón a 30°, añadir con la pipeta de Mohr, 5 ml. del producto, en unos 10 ó 12 segundos, mientras se agita continuamente con una varilla de vidrio de 4 a 6 mm. de diámetro, a cuatro vueltas por segundo, aproximadamente. La punta de la pipeta se introduce 2 cm. por debajo del borde del vaso, dirigiendo el líquido hace el centro Completar a 100 ml. con agua dura patrón, agitando continuamente, y pasar a una probeta ele 100 mililitros perfectamente limpia y seca. Dejar en reposo a 30° (± 1°) durante una hora y observar si se produce separación de liquido oleoso, o crema, en la superficie o en el fondo. En caso afirmativo no deberá exceder de 2 ml.
1.4.2. Aplicable a los productos emulsionables en proporción igual o superior al 5 por 100 y para cuya preparación debe agregarse el agua sobre ellos.
Poner en el vaso de 250 ml., 5 ml. de producto, añadiendo cantidad suficiente de agua dura patrón a unos 30°, hasta completar 100 ml., por medio de un embudo de llave. Esta adición se hará en unos 5 a 6,5 minutos, mientras se agita continuamente con una varilla de vidrio de 4 a 6 mm. de diámetro a una velocidad aproximada de cuatro vueltas por segundo. Pasar inmediatamente la emulsión a una probeta de 100 ml. perfectamente limpia y seca, dejar en reposo 1 hora a 30° (± 1°) y observar si hay separación de liquido oleoso' o crema en la superficie o en el fondo. En caso afirmativo no deberá exceder de 2 ml.
1.4.3. Cuando los productos se empleen a concentraciones considerablemente inferiores al 5 por 100, hacer las pruebas siguiendo las anteriores indicaciones, pero empleando la proporción correspondiente del producto ensayado. En caso de producirse separación de líquido oleoso o cremoso, no deberá tener un volumen superior al 40 por 100, aproximadamente, del que ocupaba el producto empleado.
1.5. Referencias.
1. O. M. S., «Specifications for Pesticides», 3.ª ed. (1967), M/13, p. 284.
2. F. A. O., «Boletín Fitosanitario» (38), 16-67 (1968).
3. F. A. O., «Boletín Fitosanitario» (Ap. 4), 11-67 (1963).
4. Ruiz Castro, A., «Bol. Pat. Veg. y Ent. Agric.». 27-295-329 (1964).
2. FINURA DE POLVOS
2.1. Principio.
Tamizado del producto, mantenido previamente en condiciones que simulan el efecto del almacenamiento.
Para aplicación a las formulaciones de plaguicidas usadas en espolvoreos.
2.2. Material y aparatos.
2.2.1. Vaso de precipitados, de vidrio fino, de 250 ml. y 60 a 65 mm. de diámetro interior.
2.2.2. Aparato de tamizado por vibración de 250 a 300 r. p. m., con tamiz de 100 µ (0,1 U. N. E.) provisto de tapas superior e inferior que ajusten sin pérdidas.
2.2.3. Disco de plomo de 56 mm. de diámetro con un peso de 625 g. (25 g/cm2). Puede usarse un recipiente de vidrio, con fondo plano, lastrado con perdigones.
2.3. Procedimiento.
Poner 20 g de la muestra en el vaso y nivelar, sin comprimir. Poner encima el disco de plomo y mantener en estufa, a 54° ± 1° durante 24 horas. Quitar el disco y dejar enfriar una hora.
Pasar el tamiz limpio y seco, cubrirlo y fijarlo en el aparato tamizador, en el que se agita 10 minutos. Desmenuzar los terrones blandos que puedan quedar, pasando suavemente un cepillo sobre el tamiz, cubrirlo y poner en marcha de nuevo. La operación se da por terminada cuando al agitar, a mano, el tamiz sobre un papel satinado, solamente pasa una cantidad insignificante de sustancia.
Pesar el residuo que queda retenido en el tamiz.
2.4. Cálculo.
El peso del residuo en gramos multiplicado por cinco nos dará el porcentaje de residuo.
2.5. Observaciones.
Para las formulaciones que contienen malation, dimetoato, o algún otro producto que pueda alterarse o cambiar sus propiedades a 54°, hacer la simulación de almacenamiento a 34° ± 1° durante 15 día.
2.6. Referencias.
1. O. M. S., «Specifications for Pesticides», M/4, 3.ª Ed. (1967), 253-254.
2. F. A. O., «Boletín Fitosanitario» (13),l0-110 (1962).
3. Ruiz Castro, A., «Bol. Pat. Veg, y Ent. Agrio.», 27-295-392 (1964).
4. Instituto de Racionalización del Trabajo, «Una Norma Española 7.050».
3. SUSPENSIBILIDAD DE POLVOS
3.1. Principio.
Obtención de la suspensión en condiciones definidas, reposo y decantación de los 9/10 superiores de volumen total, filtración, lavado y secado del residuo.
Para aplicación a las formulaciones en polvo, empleadas en forma de suspesiones acuosas.
3.2. Material y aparatos.
3.2.1. Cápsula de porcelana, de fondo redondo, de aproximadamente 250 mm. de diámetro.
3.2.2. Probeta de 500 ml., de 50 mm. de diámetro interior.
3.2.3. Sifón de tubo de vidrio.
3.2.4. Crisol de vidrio de fondo filtrante, porosidad 3.
3.2.5. Matraz kitasato con pieza de adaptación para crisol filtrante.
3.2.6. Trompa o bomba de vacío.
3.3. Reactivos.
3.3.1. Agua dura patrón. Preparar como en 1.3.
3.4. Procedimiento.
Pesar, con tres cifras exactas, la cantidad de muestra necesaria para preparar 500 ml. de dispersión, a la concentración máxima a que se use el producto.
Poner la muestra en la cápsula de porcelana, limpia y seca, y agregar un volumen de agua dura patrón doble del peso de la muestra (ml/g.). Remover enérgicamente la papilla que se forma, para deshacer los grumos que se pueden formar, durante un tiempo máximo de 5 minutos. Agregar otros 25 ml. de agua dura patrón y remover al mismo ritmo fuerte, durante 10 minutos, añadiendo a continuación durante otros 3 minutos y sin dejar de remover, el resto de agua dura patrón hasta completar los 500 ml. Inmediatamente verter el contenido en la probeta.
Dejar en reposo 30 minutos y decantar por sifonado los 450 ml. superiores, bajando el sifón lentamente para no remover el posible sedimento. Filtrar por el crisol, previamente desecado y tarado, acoplado al kitasato, los 50 ml. restantes, arrastrando y lavando el sedimento, si lo hay, con varias porciones de agua dura. Dejar el crisol escurrido, durante una noche en estufa a 54° ± 1°, enfriar en desecador y pesar.
Restando del peso obtenido la tara del crisol, se obtiene el peso del sedimento más un décimo del producto en suspensión.
3.5. Cálculo.
El resultado se expresará en tanto por ciento de la muestra empleada.
3.6. Observaciones.
Deberán tenerse en cuenta las instrucciones para preparar las suspensiones de cada producto, si difieren notablemente de éstas.
3.7. Referencias.
1. O. M. S., «Specifications for Pesticides», 3.ª Ed. (1967), M/2, 249.
2. O. M. S., «Specifications for Pesticides». 3.ª Ed. (1967), SIF/1, R3-80-81.
3. F. A. O., «Boletín Fitosanitario» (Ap. 4), 11-67 (1963).
4. Ruiz Castro, A., «Bol. Pat. Veg. y Ent. Agric.», 27-295-329 (1964).
4. DETERMINACION DE LA HUMEDAD
4.1. Principio.
Valoración del contenido en agua por desecación a 110°, para aquellos productos cuyos demás componentes no son muy volátiles a esa temperatura.
4.2. Material y aparatos.
4.2.1. Pesasustancias con tapón esmerilado, de unos 30 mm. de diámetro.
4.2.2. Cápsula de porcelana fondo plano, de unos 80 mm. de diámetro.
4.2.3. Estufa de desecación para 110°.
4.2.4. Baño de agua.
4.3. Procedimiento.
4.3.1. Productos pulverulentos.
Pesar en un pesasustancias tarado, 2 g. exactos, o una cantidad aproximada M, e introducirlo destapado en la estufa mantenida a 105-110° durante una hora. Dejar enfriar 30 minutos en desecador y pesar tapado. Volver a mantener destapado 30 minutos en la estufa, enfriar y pesar, hasta que dos pesadas consecutivas difieran en menos de 0,005 g., hallando la diferencia P entre esta pesada y la primera.
4.3.2. Productos en pasta.
Pesar en cápsula de porcelana tarada 50 g., o una cantidad aproximada M, de la pasta bien mezclada; concentrar por evaporación en baño de agua y luego desecar en estufa a 105-110° durante una hora y después en períodos sucesivos de 30 minutos, enfriando y pesando cada vez hasta diferencia menor de 0,005 g., anotando la diferencia P entre esta pesada y la inicial. La sustancia que ha sufrido esta primera desecación parcial se pulveriza, se pesan 2 g. o una cantidad aproximada M', en un pesasustancias con tapón esmerilado, tarado, con el que se procede como en el caso de productos pulverulentos, obteniendo la correspondiente pérdida de peso P'.
4.4. Cálculos.
La humedad en tanto por ciento será:
4.4.1. H = 100 P/M; si M = 2 g., H = 50 P.
4.4.2.

si M = 50 g. y M' = 2 g., H = P (2 — P') + 50 P’.
4.5. Observaciones.
En los productos conocidos, dejar inicialmente en la estufa el tiempo determinado en análisis anteriores, enfriar, pesar y desecar otros 30 minutos, comprobando que la pérdida de peso es menor que 0,005 g.
En los productos a los que no es aplicable esta norma hay que emplear otros métodos adecuados a sus propiedades (desecación en vacío sobre deshidratantes, arrastre con vapor de disolventes o determinación química del agua).
Si interesa referir las restantes determinaciones a producto seco, multiplicar por 100/100 — H los valores referidos al producto sin desecar.
4.6. Referencias.
1. O. M. S., «Specifications for Pesticides», 3.ª Ed. (1907), M/8, 270-271.
2. F. A. O., «Boletín Fitosanitario» (10), 10-39 (1962).
3. Ruiz Castro, A., «Bol. Pat. Veg. y Ent. Agric.», 27-295-392 (1964).
4. A. O. A. C,, «Official Methods of Analysis», 10.ª Ed. (1965), 4.003.
Métodos de análisis para la determinación de las características de los azufres destinados a usos fitosanitarios
FINURA (TAMIZACION SECA)
1. Principio.
Tamizado por vibración a través del tamiz o tamices adecuados y pesada de la porción rechazada.
2. Material y aparatos.
Juego de tamices, de mallas adecuadas, con tapa y recipiente. Vibrador de tamices (más dj 250 vibraciones minuto).
Cápsula de porcelana de unos 100 mm. de diámetro.
3. Procedimiento.
Pesar 50 g. de la muestra con error menor de 0,1 g. y pasarla al tamiz, limpio y secó, encajado en el recipiente. Tapar y adaptar el vibrador, que se pone en funcionamiento 10 minutos. Parar, destapar y observar si sobre el tamiz quedan terrones blandos, y en caso afirmativo, desmenuzarlos pasando suavemente un cepillo sobre el tamiz y volver a vibrar, tapado, otros 10 minutos.
Pasar a una cápsula tarada el polvo que quedó en el tamiz y pesar con error menor de 0,01 g.
4. Cálculo.
Calcular el porcentaje de rechazo para el tamiz usado:
% rechazo = 2 P.
P = peso del polvo que quedó en el tamiz.
5. Observaciones.
El juego de tamices debe de estar realizado en forma que la tapa y cada uno de los bastidores de los tamices puedan encajarse con ajuste suave y sin pérdidas entre sí, y en el recipiente. Todas las soldaduras deben ser redondas y pulidas, sin ángulos ni poros donde pueda quedar retenido el polvo.
INSOLUBILIDAD EN SULFURO DE CARBONO
1. Principio.
Disolución de la fracción cristalina de los azufres por tratamiento con sulfuro de carbono en extractor continuo, para deducir por diferencia la fracción amorfa insoluble, presente en los azufres sublimados puros.
2. Material y aparatos.
Aparato extractor Soxhlet, pequeño.
Cartuchos de papel de filtro.
Baño de agua eléctrico.
Estufa a 70-80°.
3. Reactivos.
Sulfuro de carbono para análisis. No debe dejar residuo apreciable por evaporación.
4. Procedimiento.
Pesar 10 g. de la muestra en un cartucho de papel de filtro que se coloca en el aparato Soxhlet, cuyo matraz se ha tarado previamente y añadirle suficiente sulfuro de carbono para conseguir el sifonado y unos 50 ml. más.
Calentar el matraz tarado del aparato en el baño de agua, de forma que sifone el cuerpo extractor cada dos o tres minutos, durante una hora. Evaporar el extracto con precaución, sobre baño de agua y en vitrina con buen tiro. Desecar a 70-80°, enfriar en desecador y pesar.
5. Cálculo.
Calcular en porcentaje la porción insoluble:
% insoluble = 10 (P — P1)
P = peso de la muestra.
Pi = peso del extracto seco.
6. Observaciones.
Se deben extremar las precauciones en él manejo del sulfuro de carbono (vapores tóxicos y explosivos), especialmente observando que el refrigerante del extractor tenga siempre agua abundante y evitando la proximidad de llamas o chispas al evaporarlo.
RIQUEZA
1. Principio.
Disolución del azufre en solución de sosa, en caliente, y oxidación para transformarlo en ion sulfato, que se determina gravimétricamente como sulfato bárico.
2. Material y aparatos.
Vaso de precipitados de vidrio fino, de 600 ml.
Vidrio de reloj, de diámetro algo superior al del vaso.
Pipetas de Mohr de 5 ml. y 25 ml.
Probetas de 30 ml.
Crisol de vidrio poroso G-4.
Estufa graduada a 130°.
Matraz Kitasato de 500 ml. con pieza para adaptar el crisol.
3. Reactivos.
Disolución de hidróxido sódico para análisis al 30 por 100 (d = 1,33).
Agua oxigenada para análisis al 30 por 100 (110 vol.).
Solución de ácido clorhídrico 1:3. Diluir el concentrado (d = 1,19) con un volumen triple de agua destilada.
Solución de cloruro bárico para análisis al 10 por 100.
Solución alcohólica de naranja de metilo al 0,1 por 100.
4. Procedimiento.
Pesar en un vidrio de reloj, limpio y seco, unos 200 mg. dé producto, con precisión de 0,1 mg., y pasarlos por arrastre con unos 20 ml. de agua destilada a un vaso de vidrio fino de 600 ml.
Añadir 5 ml de sosa al 30 por 100 y calentar hasta conseguir la disolución total de la muestra, y a continuación diluir con unos 100 ml de agua destilada y 25 ml. de agua oxigenada al 30 por 100, tapar con el vidrio de reloj y calentar suavemente a ebullición, que se mantendrá hasta que cese el desprendimiento de gas (unos treinta minutos).
Lavar con agua destilada el vidrio de reloj, sobre el vaso, añadir dos gotas de indicador naranja de metilo, neutralizar con ácido clorhídrico 1:3 y añadir un exceso de unos 3 ml.
Hervir y precipitar en caliente y agitando, con 115 ml. de cloruro bárico al 10 por 100. Dejar reposar treinta minutos y filtrar el líquido caliente por el crisol de vidrio G-4 previamente limpio, seco a 130° y tarado.
Lavar con agua destilada caliente hasta que el filtrado esté exento de iones bario. Suele bastar con tres lavados.
Secar a 130° dos horas, enfriar en desecador y pesar.
Simultáneamente se realizará una valoración en blanco con los reactivos para determinar su contenido en azufre.
5. Cálculo.
Calcular el porcentaje de azufre en la muestra:

P1 = SO4Ba de la muestra.
P2 = SO4Ba del ensayo en blanco.
P = peso de la muestra.
ACIDEZ TOTAL
1. Principio.
Extracción de los ácidos con agua destilada y valoración con hidróxido sódico.
2. Material y aparatos.
Vidrio de reloj.
Matraz erlenmeyer de 250 ml. con tapón esmerilado.
Matraz aforado de 200 ml.
Pipeta de 100 ml.
Vaso de precipitados de 200 ml.
Bureta.
3. Reactivos.
Papel tornasol azul.
Indicador naranja de metilo (solución alcohólica al 0.1 por 100).
Hidróxido sódico 0,1 N.
4. Procedimiento.
Pesar 10 g. de muestra, con error menor de 0,01 g., y transvasar con unos 100 ml. de agua destilada al matraz erlenmeyer Tapar y agitar enérgicamente durante dos minutos y a continuación filtrar al matraz aforado de 250 ml. y lavar el matraz erlenmeyer y el filtro con pequeñas porciones de agua destilada hasta que el filtrado no enrojezca el papel de tornasol azul.
Completar el volumen, agitar bien, tomar 100 ml., que se valoran en el vaso, previa adición de dos gotas de indicador naranja de metilo, con la solución de hidróxido sódico 0,1 N.
5. Cálculo.
Calcular el porcentaje de acidez, expresado en ácido sulfúrico:
% acidez total = 0,1225 × V g.
V = volumen de solución de hidróxido sódico, exactamente 0,1 N.
ANHIDRIDO SULFUROSO LIBRE
1. Principio.
Valoración por iodometría del líquido preparado para la valoración de la acidez total. Solamente se determina en los azufres sublimados si la acidez total fuese mayor de 0,46 por 100, expresado en ácido sulfúrico, equivalente al 0,3 por 100 de SO2 admisible.
2. Material y aparatos.
Pipeta de dos enrases, de 2 ml.
Vaso de precipitados, de 100 ml.
Bureta.
3. Reactivos.
Solución de iodo 0,1 N.
Ioduro potásico para análisis.
Indicador de almidón: Desleír bien 1 g. de almidón soluble en unos 10 ml. de agua destilada y añadir, agitando, sobre 100 ml. de agua hirviendo y continuar la ebullición dos minutos. Enfriar y conservar bajo una ligera capa de tolueno; tomar con pipeta.
4. Procedimiento.
Poner en el vaso 2 ml. de solución de iodo 0,1 N y añadir lentamente, con bureta, el líquido filtrado preparado para valorar la acidez total. Cuando el color sea ligeramente amarillo, añadir un cristalito de ioduro potásico y 3 ml. de indicador de almidón y continuar la adición hasta que desaparezca el color azul.
5. Cálculo.
Calcular el porcentaje de anhídrido sulfuroso libre

V = volumen de líquido usado.
CENIZAS
1. Principio.
Valoración de las cenizas residuales de calcinar a 800.º
2. Material y aparatos.
Crisol de cuarzo.
3. Procedimiento.
Pesar unos 5 g. de muestra en crisol de cuarzo, previamente calcinado a 800°, enfriando en desecador y tarado.
Quemar en vitrina de gases con mechero de Bunsen y calcinar en horno a 800° durante una hora. Enfriar en desecador y pesar.
4. Cálculo.
Calcular el porcentaje de cenizas:

p = aumento de peso del crisol.
p = peso de la muestra.
HUMEDAD
1. Principio.
Determinar la pérdida de peso a 90-95°.
2. Material y aparatos.
Pesafiltros con tapón esmerilado.
Estufa a 90-95°.
3. Procedimiento.
En un pesafiltros limpio, seco y tarado pesar unos 10 g. de la muestra y mantener a 90-95° hasta peso constante.
4. Cálculo.
Calcular el porcentaje de humedad:

p = pérdida de peso.
p = peso de la muestra.
SUSPENSIBILIDAD
1. Principio.
Preparación de una suspensión que se deja sedimentar y se decanta, determinando el aumento en sólidos de la porción inferior.
2. Material y aparatos.
Vidrio de reloj.
Probeta graduada a 250 ml., cor. tapón esmerilado.
Baño de agua eléctrico.
Estufa a 105°.
3. Productos.
Alcohol etílico 96 por 100 v/v. No debe dejar residuo apreciable por evaporación.
4. Procedimiento.
Pesar 1,25 g. de la muestra y pasar a la probeta. Agitar hasta suspensión uniforme con unos 200 ml. de agua destilada y completar el volumen a los 250 ml.
Dejar reposar cinco minutos,, invertir la probeta diez veces y dejar reposar treinta minutos.
Sifonar los 225 ml. superiores y pasar los 25 ml. restantes a la cápsula tarada, así como varios lavados con alcohol hasta que no quede residuo sólido en la probeta.
Evaporar a sequedad sobre baño de agua hirviente, secar en estufa treinta minutos, dejar enfriar en desecador y pesar.
5. Cálculo.
Calcular en porcentaje de suspensibilidad:

P = peso de la muestra.
r = residuo en la cápsula.
1. GRADO DE FINURA
1.1. Principio.
Deshacer los agregados originados por simple comprensión mecánica, tamizar y calcular el porcentaje que pasa por el tamiz (grado de finura), con el fin de comprobar si se ajusta a las disposiciones vigentes en la materia.
Aplicable a los fertilizantes y productos afines sólidos, de acuerdo con las especificaciones granulométricas que en cada caso son exigibles.
1.2. Material y aparatos.
1.2.1. Juego de tamices, con las siguientes aberturas de malla: 25 mm., 6,3 mm., 4,0 mm., 2,0 mm., 1,0 mm., 0,5 mm., 0,6 mm. (para escorias), 0,16 mm. (para escorias), 0,125 mm. (para fosfato de roca), 0,063 mm. (para fosfato de roca). Norma UNE-7.050.
1.2.2. Rodillo de caucho duro, taco de madera o utensilio similar.
1.3. Procedimiento.
Tomar unos 50 g. del material contenido en el frasco, por cuarteado, y deshacer los agregados con un rodillo de caucho duro, taco de madera u otro utensilio adecuado para desagregar sin triturar las partículas.
Tamizar el material disgregado, pesando lo que queda en el tamiz y. lo que pasa.
1.4. Cálculo.
El grado de finura G será:

siendo:
P = peso del fertilizante que pasa por el tamiz.
p = peso del fertilizante que queda sobre el tamiz.
1.5. Referencias.
1. Ministerio de Agricultura. Orden ministerial de 10 de junio de 1970.
2. A. O. A. C., «Official Methods of Analysis», 1.002. Washington, 1970.
2. PREPARACION DE LA MUESTRA PARA EL ANALISIS
2.1. Procedimiento.
Salvo que en el método correspondiente de análisis se indique otra cosa, preparar la muestra para análisis de la forma siguiente:
Pasar la totalidad del contenido, triturar con un mortero de mane y pasar por un tamiz de 0,5 mm. de abertura de mallas. Si los fertilizantes están húmedos, pasar por un tamiz de 1 mm, en lugar del de 0,5 mm.
Las partes gruesas que quedan en el tamiz y que por su dureza no son triturables se pesarán y se tendrán en cuenta, haciendo constar su tanto por ciento referido al peso total de la muestra, para dar un porcentaje definitivo en elementos fertilizantes efectivos.
La trituración se hace lo más rápidamente posible para impedir pérdidas o ganancias de humedad durante la operación.
Mezclar bien y guardar la muestra en frascos herméticamente cerrados.
En los casos en que la muestra lo permita se puede utilizar un molinillo eléctrico, evitando calentamiento excesivo, y usando la cantidad necesaria para que se realice una óptica molienda.
2.2. Referencias.
1. Ministerio de Agricultura. Orden ministerial de 10 de junio sobre ordenación y control de productos fertilizantes y afines. Madrid, 1970.
2 A. O. A. C., «Official Methods of Analysis», 2.007. Washington, 1970.
DETERMINACION DE LA HUMEDAD
3. AGUA TOTAL (METODO DE DESECACION EN ESTUFA)
(No aplicable a muestras que producen sustancias volátiles diferentes de H2O a la temperatura de desecación)
3.1. Procedimiento.
Calentar 2 g. de la muestra debidamente preparada para análisis durante cinco horas en una estufa a 99-101 °C. En el caso de NO3Na. SO4(NH1)2 y sales de K, calentar a 129-131 °C. Expresar el porcentaje de pérdida en peso a la temperatura utilizada como contenido de agua.
3.2. Referencias.
1. A. O. A. C., «Official Methods of Analysis», 2.012. Washington, 1970.
4. AGUA LIBRE (METODO DE DESECACION CON VACIO)
4.1. Procedimiento.
Poner 2 g. de la muestra debidamente preparada para análisis en pesa-filtro tarado (si se trata de materias extremadamente higroscópicas o húmedas, pesar por diferencia en pesafiltros tapados). Desecar la muestra a 25-30 °C (para obtener resultados exactos, la temperatura debe ser lo más constante posible) en desecador de vacío sobre (ClO4)2Mg. anhidro, P2O5 o BaO, con un vacío comprendido entre 500 y 560 mm. (200-250 mm. de presión absoluta) durante dieciséis-dieciocho horas. Volver a pesar y referir el porcentaje de pérdida en peso como H2O libre.
4.2. Referencias.
1. A. O. A. C., «Official Methods of Analysis», 2.013. Washington, 1970.
5. DETERMINACION DEL NITROGENO (DETECCION DE NITRATOS)
5.1. Principio.
El óxido nítrico producido por la reacción del ácido nítrico con sales ferrosas, en ácido sulfúrico concentrado, se une a un exceso de sal ferrosa originando un complejo de color pardo.
5.2. Material y aparatos.
5.2.1. Tubos de ensayo.
5.3. Reactivos.
5.3.1. SO4H2 concentrado exento de N.
5.3.2. SO4Fe. 7H2O (disolución saturada).
5.3.3. NO3Na (disolución al 1 por 100),
5.4. Procedimiento.
Tratar 5 g. de la muestra con 25 ml. de agua caliente y filtrar.
A un volumen del filtrado añadir dos volúmenes de SO4H2 concentrado y dejar enfriar. Añadir unas gotas de la disolución de SO4Fe de modo que no se mezclen los líquidos.
Si hay nitratos, en la zona de unión de ambos líquidos aparece un anillo púrpura que pasa a marrón. Si hay poco NO3 da color rojizo.
A otra porción del filtrado añadir 1 ml. de la disolución de nitrato sódico y repetir el ensayo para comprobar si se agregó suficiente ácido sulfúrico la primera vez.
5.5. Observaciones.
Esta determinación previa es indispensable para saber qué método hay que elegir para la determinación del N total, salvo que se use el método comprensivo.
5.6. Referencias.
1. «Método A. O. A. C.» Edición de 1970, número 2.048.
6(a). NITROGENO TOTAL
(Método de Kjeldahl modificado para muestras que no contengan nitratos)
6(a).1. Principio.
Transformar el nitrógeno orgánico en sulfato amónico, por ebullición con SO4H2 concentrado, y separar por destilación el conjunto de nitrógeno amoniacal así formado y el que eventualmente pudiera existir en la muestra, recogiéndolo en un exceso de ácido valorado. El exceso de ácido se determina por retorno con un álcali valorado en presencia de rojo de metilo.
Es aplicable a todos los abonos que no contengan nitrato.
6(a).2. Material y aparatos.
6(a).2.1 Matraces Kjeldahl.
6(a).2.2. Aparatos de destilación.
6(a).2.3. Vasos de 300 ml.
6(a).2.4. Buretas de 25 ml.
6(a).2.5. Probetas de 25 y 200 ml.
6(a).3. Reactivos.
6(a).3.1. Oxido de mercurio o mercurio metálico, exento de N.
6(a).3.2. Sulfato potásico o sódico anhidro, exento de N.
6(a).3.3. Acido sulfúrico de 93 a 98 por 100, exento de N.
6(a).3.4. Disolución de tiosulfato sódico o de sulfuro sódico: 80 g. de S2O3Na2 ⋅ 5H20 en 1 litro de agua o 40 g. de sulfuro sódico en 1 litro de agua.
6(a).3.5. Hidróxido sódico sólido o en disolución: 450 g. de NaOH en agua, enfriar y enrasar a 1 litro. Debe tener de densidad 1,36 o más.
6(a).3.6. Granalla de zinc.
6(a).3.7. Polvo de zinc impalpable.
6(a).3.8. Rojo de metilo: Disolver 1 g. en 200 ml. de alcohol.
6(a).3.9. Disolución de ácido sulfúrico o clorhídrico N/2, o N/10 si la cantidad de N es pequeña.
6(a).3.10. Disolución de sosa N/2 o N/10.
6(a).4. Procedimiento.
6(a).4.1. Pesar de 0,7 a 2,2 g. del abono y ponerlos en un matraz Kjeldahl. Añadir 0,7 g. de óxido mercúrico o 0,05 g. de mercurio metálico, 15 g. de sulfato potásico o sódico anhidro y 25 ml. de sulfúrico concentrado. Si fuera necesario pesar más de 2,2 g., añadir lo ml. de sulfúrico por cada g. de muestra.
Colocar el matraz en posición inclinada y calentar suavemente hasta que cese la formación de espuma (para reducir ésta puede añadirse una pequeña cantidad de parafina). Hervir vivamente hasta que la solución se aclare y luego, por lo menos, otros treinta minutos más (dos horas para las muestras que contengan materia orgánica).
6(a).4.2. Enfriar, añadir con precaución unos 200 ml. de agua, volver a enfriar por debajo de 25 °C, añadir 25 ml. de disolución de tiosulfato y mezclar para precipitar el Hg. Añadir unos gránulos de Zn. para evitar vaporación súbita, inclinar el matraz y añadir sin agitar la sosa (25 g. sólidos o suficiente disolución para poner la reacción muy alcalina). La disolución de tiosulfato o sulfuro se puede mezclar con la disolución de sosa antes de añadirla al matraz.
6(a).4.3. Inmediatamente conectar el matraz al bulbo teniendo sumergido el extremo de la alargadera en un vaso que contiene el ácido o,5N o 0,1N, exactamente medidos, y cinco a siete gotas del indicador. Agitar el matraz para mezclar el contenido y calentar hasta que destile todo el amoniaco (al menos 150 ml. de destilado).
6(a).4.4. Valorar el exceso de ácido con. sosa de la misma normalidad (A ml. de sosa).
Hacer un ensayo en blanco (B ml. de sosa).
6(a).5. Cálculo.

P = peso, en g., de la muestra.
A = volumen, en ml., de sosa 0,5N consumido en el análisis.
B = volumen, en ml., de sosa 0,5N consumido en el ensayo en blanco.
6(a).6. Observaciones.
6(a).6.1. El SO4H2 hay que añadirlo siempre sobre el agua.
6(a).6.2. Usar protectores de cara y manos (guantes, etc.).
6(a).6.3. Las sales de Hg. son muy tóxicas y casi todas solubles en agua. Emplear protección de piel y vías respiratorias.
6(a).6.4. El Hg. es peligroso en contacto con el amoniaco, halógenos y álcalis. Los vapores son muy tóxicos y acumulativos. Si se derrama sobre superficies calientes es muy peligroso; limpiarlo rápidamente. Para ayudar a la limpieza, espolvorear sobre el Hg. azufre en polvo. Cuando se evapora hay que hacerlo en campana de buen tiro.
6(a).6.5. Para impedir la contaminación del medio, diluir el líquido que queda en los Kjeldahl con agua 1 + 1, separando por filtración las sales de Hg. insolubles que se reservan en un recipiente apropiado.
6(a).6.6. Para la digestión, utilizar Kjeldahls de vidrio duro moderadamente grueso y bien recocido de 500 a 800 ml. La prueba del dispositivo de calentamiento se hace ajustándolo para llevar 250 ml. de agua a 25 °C a ebullición fuerte en unos cinco minutos o el tiempo que especifique el método. Para probar los calentadores se precalientan diez minutos sí son de gas o treinta minutos si son eléctricos. Agregar tres o cuatro granallas para evitar sobrecalentamiento.
6(a).7. Referencias.
1. «Métodos oficiales, A. O. A. C.» Edición 1970, números 2.049 y 2.051.
6(b). NITROGENO TOTAL
(Método de Kjeldahl modificado para muestras que contienen nitratos)
6(b).1. Principio.
El nitrato nitra el ácido salicílico en presencia de ácido sulfúrico concentrado. Posteriormente, el nitrógeno del derivado nitrato se reduce por el Zn. o el tiosulfato y se prosigue como en el Kjeldahl para muestras que no contengan nitratos.
No es aplicable a abonos líquidos o sólidos que tengan alta relación Cl/NO3.
6(b).2. Material y aparatos.
Como en 6(a).2.
6(b).3. Reactivos.
6(b).3.1. Acido salicílico y los empleados en 6(a).3.
6(b).4. Procedimiento.
6(b).4.1. Colocar de 0,7 a 2,2 g. de muestra en el matraz de digestión y añadir 40 ml. de SO4H2 que contenga 2 g. de ácido salicílico. Agitar hasta mezclarlo completamente y dejar en reposo agitándolo de cuando en cuando durante treinta minutos o más.
6(b).4.2. Añadir 5 g. de S2O2Na2. 5H20 o 2 g. Zn. en polvo. Agitar y dejar en reposo cinco minutos. Calentar con llama pequeña hasta que cese la formación de espuma. Retirar del fuego, añadir 0,7 g. de HgO o 0,65 g. de Hg. metálico y 15 g. de sulfato potásico o sódico anhidro. Hervir vivamente hasta que se aclare la disolución y luego treinta minutos más por lo menos (dos horas si la muestra contiene materia orgánica).
6(b).4.3. Continuar hasta el final como en 6(a).4.2 y siguientes.
6(b).5. Referencias.
1. «Métodos oficiales, A. O. A. C.» Edición 1970, número 2.052.
6(c). NITROGENO TOTAL (Método comprensivo)
6(c).1. Principio.
La reducción del nitrógeno nítrico a amoniacal se hace por el cromo en polvo, prosiguiéndose como en el Kjeldahl para muestras sin nitratos.
Es aplicable a todas las formas de nitrógeno.
6(c).2. Material y aparatos.
Como en 6(a).2.
6(c).3. Reactivos.
6(c).3.1. Cromo metal en polvo que pase por un tamiz de 100 mallas (0,149 milímetros), exento de N.
6(c).3.2. Alundum.
6(c).3.3. Acido sulfúrico diluido. Se agregan lentamente 625 ml. de SO4H3 a 300 ml. de agua. Diluir a casi un litro con agua y agitar. Enrasar cuando se enfría. Avitar presencia de amoníaco en la atmósfera.
6(c).3.4. Disolución de tiosulfato. sódico o de sulfato potásico: 100 g. de S2O2Na2 5H2O disolver en agua y enrasar a un litro, o 30 g. de SK2 en un litro de agua.
6(c).3.5. Los indicados en 6(a).3.
6(c).4. Procedimiento.
6(c).4.1. Pesar de 0,2 a 2 g. de la muestra (que contenga una cantidad igual o menor de 00 mg.- de N. nítrico), ponerlos en un Kjeldahl de 500 a 800 ml. y agregar 1,2 g. de polvo de cromo. Agregar 35 ml. de agua, o si el problema es líquido, completar hasta formar un volumen total de 35 ml. Dejar en reposo durante diez minutos agitando en circulo de cuando en cuando suavemente para disolver todos los nitratos. Agregar 5 ml. de ClH y dejar en reposo un tiempo que no debe ser inferior a treinta segundos ni superior a diez minutos.
6(c).4.2. Poner el matraz en un mechero precalentado con una energía calorífica suficiente para que lo haga hervir en siete o siete minutos y medio. Después de calentar tres minutos y medio, separar del calor y dejar enfriar. Agregar 22 g. de sulfato potásico, 1 g. de HgO y unos gránulos de alundum. Añadir 40 ml. de SO4H2 diluido (si se dispone de ventilación adecuada se pueden agregar 25 ml. de SO4H2 concentrado en lugar de diluido). Si la materia orgánica, que consume gran cantidad de ácido, excede de 1 g., agregar adicionalmente 1 ml. de SO4H2 concentrado por cada 0,1 g. de materia orgánica en exceso.
6(c).4.3. Colocar el matraz en un mechero, precalentando y elevar después la temperatura para que hierva en cinco minutos (los mecheros precalentados reducen la formación de espuma en la mayor parte de las muestras).
Disminuir el calor si la espuma ocupa una zona igual o mayor de 2/3 del bulbo del matraz. Regular el calor hasta que pase esta fase. Calentar en el citado mechero durante cinco minutos hasta que los humos blancos y densos del sulfúrico aclaren el bulbo del matraz. La digestión es ahora completa para muestras que contienen nitrógeno amoniacal, nítrico y ureico. Para otras muestras, agitar suavemente por rotación y proseguir la digestión durante sesenta minutos más.
6(c).4.4. Continuar hasta el final como en 0(a).4.2 y siguientes.
6(c).5. Referencias.
1. «Métodos Oficiales A.O.A.C.» Edición 1970, números 2.051, 2.053 y 2.054.
7(a). NITROGENO AMONIACAL (Método del óxido de magnesio)
7(a).1. Principio.
Transformar el nitrógeno amoniacal en amoníaco por la acción de MgO (no carbonatado). Destilar el amoníaco sobre un volumen conocido de SO4H2 N/2 y valorar el exceso do ácido por retorno con NaOH N/2 en presencia de rojo de metilo.
Aplicable a todos los abonos, incluso los compuestos, en los cuales el nitrógeno se encuentre sólo como sal amónica o mezclada con nitratos.
No es aplicable a los abonos que contengan urea, cianamida y otros compuestos orgánicos nitrogenados.
7(a).2. Material y aparatos.
Como en 6(a).2, excepto matraz Kjeldahl.
7(a).3. Reactivos.
7(a).3.1. MgO (libre de carbonato magnésico).
7(a).3.2. NaOH N/2.
7(a).3.3. SO4H2 N/2.
7(a).3.4. Rojo de metilo. Un g. en 200 ml. de alcohol.
7(a).4. Procedimiento.
7(a).4.1. Colocar de 0,7 a 3,5 g. de la muestra, según el contenido de amoníaco, en un matraz de destilación con unos 200 ml. de agua y 2 g. o más de MgO. Conectar el matraz a un refrigerante mediante un bulbo de seguridad.
7(a).4.2. Destilar unos 100 ml. de líquido y recoger en un vaso conteniendo unos 30 ml. de sulfúrico N/2 y unas gotas de rojo de metilo. Valorar el exceso de sulfúrico N/2 mediante sosa N/2 (A ml. de sosa). Hacer un ensayo en blanco (B ml. de sosa).
7(a).5. Cálculo.
Como en 6 (a).5.
7(a).6. Referencias
1. «Métodos de análisis A.O.A.C.» Edición 1970, número 2.057.
7(b). NITROGENO AMONIACAL (Método del formaldehído)
7(b).1. Principio.
El catión NH4 reacciona con el formol originando urotropina y dejando en libertad los ácidos de la sal, que se valoran.
Es necesario que la disolución de la muestra sea- neutra respecto al rojo de metilo y que no existan malcrías sólidas que puedan reaccionar con los ácidos liberados.
Es aplicable al nitrato amónico y al sulfato amónico. Puede utilizarse en presencia de urea.
7(b).2. Material y aparatos.
7(b).2.1. Matraces aforados de 250 a 500 ml.
7(b).2.2. Vasos de 300 ml.
7(b).2.3. Buretas de 25 ml.
7(b).2.4. Probetas de 200 ml.
7(b).3. Reactivos.
7(b).3.1. Formaldehído al 37 por 100.
7(b).3.2. NaOH N/2.
7(b).3.3. Fenolftaleína (disolución alcohólica al 1 por 100).
7(b).3.4. Rojo de metilo (disolución alcohólica al 0,1 por 100).
7(b).4. Procedimiento.
7(b).4.1. Pesar exactamente una cantidad próxima a 7 ó 14 gramos de la muestra y diluir a 250 ml. o a 500 ml. Pipetar 25 ó 50 ml. en un erlenmeyer y añadir 1 ml. de formaldehído por cada 0,1 g. de muestra en la alícuota. Diluir a unos 200 ml. y dejar en reposo cinco minutos.
7(b).4.2. Valorar con una solución de NaOH N/2 en presencia de cinco gotas de fenolftaleína.
7(b).4.3. Hacer, un ensayo en blanco con el formaldehído y la fenolftaleína.
7(b).4.4. Neutralizar una alícuota del problema, igual a la usada en la deformación, con NaOH N/2 en presencia del rojo de metilo.
7(b).5. Cálculo.

M = ml. de NaOH N/2 gastados en 7 (b).4.2.
A = ml. de NaOH N/2 gastados en 7(b).4.3.
B = ml. de NaOH N/2 gastados en 7(b).4.4.
P = peso de la muestra contenida en la alícuota, en g.
7(b).6. Observaciones.
Es necesario neutralizar el formaldehído y el problema en alícuotas distintas a las de la determinación, aunque iguales en volumen, para evitar la mezcla de indicadores.
7(b).7. Referencias.
1. «Métodos de análisis A.O.A.C.» Edición 1970, número 2.058.
8. NITROGENO AMONIACAL Y NITRICO CONJUNTAMENTE (Método de Devarda)
8.1. Principio.
El nitrógeno nítrico se reduce a amoniacal por el hidrógeno desprendido al reaccionar la aleación de Devarda (Al-Cu-Zn, 45/50/5) en medio fuertemente alcalino. Destilar el amoníaco formado y el ya existente en la disolución, recogiéndolo en un exceso de ácido y valorando por retorno.
Es aplicable a los abonos en que se exija el nitrógeno nítrico y amoniacal, pero no es aplicable en presencia de materia orgánica, cianamida de calcio y urea.
8.2. Material y aparatos.
Como en 6(a).2, excepto el matraz Kjeldahl.
8.3. Reactivos.
8.3.1. Aleación de Devarda.
8.3.2. Disolución concentrada de NaOH (42 por 100 en peso).
8.3-3. Disolución de SO4H2 N/2.
8.3.4. Disolución de NaOH N/2.
8.3.5. Rojo de metilo (disolución alcohólica al 0,1 por 100).
8.4. Procedimiento.
8.4.1. Pesar 0,35 a 0,5 g. de muestra y ponerlos en un matraz de 600 a 700 ml. Añadir 300 ml. de agua, 3 g. de aleación de Devarda y 5 ml. de la disolución concentrada de sosa, despacio y por las paredes para que no se mezcle con el resto.
8.4.2. Conectar con el aparato de destilación, teniendo introducido el extremo de la alargadera en un vaso que contiene 30 ml. de SO4H2 N/2. Agitar el matraz y calentar suavemente al principio y luego en una proporción que produzca 250 ml. de destilado en una hora.
8.4.3. Una vez destilado todo el amoníaco valorar el exceso de SO4H2 N/2 con sosa N/2 en presciencia de rojo de metilo (A ml. de sosa). Hacer un ensayo en blanco (B ml. de sosa).
8.5. Cálculo.
Como en 6 (a).5.
8.6. Observaciones.
Si se desea conocer separadamente el nitrógeno nítrico una vez determinados los dos juntos, determinar el nitrógeno amoniaca] por el método del óxido de magnesio y la diferencia será el nitrógeno nítrico.
8.7. Referencias.
1. Métodos de análisis A.O.A.C.» Edición 1970, número 2.060.
9. NITROGENO NITRICO (Método de Robertson)
9.1. Principio.
Determinar el N total y el N insoluble en agua. La diferencia entre ambos es el N soluble.
En la disolución de N soluble eliminar el N nítrico al estado de óxido nítrico por medio de sulfato ferroso. Una vez eliminado, determinar el N total en el residuo y la diferencia entre el N soluble y este último es el N nítrico.
Aplicable en presencia de cianamida cálcica y urea.
9.2. Material y aparatos.
Como en 6 (a).2.
9.3. Reactivos.
9.3.1. SO4Fe ⋅ 7 H20.
9.3.2. Como en 6 (a).3.
9.4. Procedimiento.
9.4.1. Modalidad A: Caso general en que es necesario determinar el N insoluble en agua.
Determinar el N total por el método Kjeldahl para muestras con nitratos, o por el método comprensivo.
9.4.2. Separar y determinen' el N insoluble en agua por el método correspondiente.
9.4.3. En la disolución obtenida en 9.4.2 eliminar el N nítrico y determinar el N restante.
Para ello, colocar el filtrado procedente del apartado anterior en un matraz Kjeldahl de 500 ml. y añadir 2 g. de SO4Fe⋅7H2O y 20 ml. de SO4H2 (si el N total es mayor del 5 por 100, poner 5 g. de SO4Fe ⋅ 7H20).
9.4.4. Ponerlo a la llama hasta evaporar el agua y aparezcan humos blancos. Continuar la digestión por lo menos diez minutos más para expulsar todo el N nítrico. Si se produce una fuerte vaporización, añadir 10 ó 15 perlas de vidrio.
9.4.5. Agregar 0,65 g. de Hg. o 0,7 de HgO, y continuar la digestión hasta que toda la materia orgánica se haya oxidado.
Enfriar, diluir y continuar como en 6(a).4.2 y siguientes.
9.4.6. Modalidad B: Modificación de Jones para el caso en que no hace falta determinar el N insoluble en agua por ser todo él soluble.
Determinar el N total por el método Kjeldahl para muestras con nitratos o por el método comprensivo.
9.4.7. Eliminar el N nítrico y determinar el N restante.
Para ello pesar 0,5 g. del problema, colocarlos en un matraz
Kjeldahl de 500 ml., añadir 50 ml. de agua y agitar suavemente.
Agregar 2 g. de SO4Fe⋅7H2O y 20 ml. de SO4H2, continuando como en 9.4.4 y 9.4.5.
9.5. Cálculo.
Modalidad A:
N total — N insoluble = N soluble.
N soluble — N obtenido en 9.4.5 = N nítrico.
Modalidad B:
N total antes de eliminar el N nítrico menos N total después de su eliminación = N nítrico.
9.6. Referencias.
1. «Método A. O. A. C.» Edición 1970, números 2.061 y 2.062.
10(a). NITROGENO UREICO (Método del xanthidrol)
10(a).1. Principio.
La urea se precipita con el xanthidrol formando dixantilurea. Después desecar y pesar el precipitado.
También se precipita el biuret, pero la cantidad existente es generalmente muy débil con relación a la de la urea, por lo que no tiene importancia en la determinación.
Aplicable a todos los abonos que contengan una cantidad de urea correspondiente a un contenido de nitrógeno superior al 3 por 100.
10(a).2. Material y aparatos.
10(a).2.1. Crisol de Gooch, con membrana filtrante de vidrio, porosidad número 4 (5-15 micras de diámetro).
10(a).2.2. Frasco de un litro.
10(a).2.3. Agitador rotativo.
10(a).2.4. Matraz aforado de 500 ml.
10(a).2.5. Pipeta de 25 ml.
10(a).2.6. Vaso de 100 ml.
10(a).3. Reactivos.
10(a).3.1. Acido acético glacial.
10(a).3.2. Disolución de xanthidrol: Preparar una solución de xanthidrol puro en etanol o metanol al 5 por 100 p/v.
10(a).3.3. Etanol de 96 por 100.
10(a).3.4. Acido acético (1 + 1): Diluir el ácido acético glacial con un volumen igual de agua.
10(a).3.5. Acido sulfúrico (1 + 50): Agregar un volumen de ácido sulfúrico concentrado a 50 volúmenes de agua.
10(a).4. Procedimiento.
10(a).4.1. Pesar 10 g. de la muestra con la aproximación de 1 mg., triturarla finamente en un mortero pequeño con algunos mililitros de agua, transfiriendo el contenido a un frasco de un litro con 400 ml. de ácido sulfúrico diluido. Agitar en un agitador rotativo a 40 vueltas por minuto durante dos horas a una temperatura de 20° ± 5 °C, filtrar sobre papel sin cenizas seco, recogiendo el filtrado en un matraz aforado de 500 ml. Lavar tres veces el residuo no disuelto por decantación y el filtro dos veces con agua. Enrasar hasta la marca y agitar.
10(a).4.2. Medir una parte alícuota de la disolución (v. ml.) que no contenga más de 20 mg de urea, colocarlos en un vaso de 100 ml. y agregar 40 ml. dé ácido acético glacial. Agitar durante un minuto con un agitador de vidrio. Si se forma un precipitado se deja el vaso en reposo durante cinco minutos filtrando el contenido. Tirar el precipitado después de haber lavado el filtro con unas gotas de ácido acético diluido. Agregar el líquido del lavado al filtrado para proseguir el análisis.
10(a).4.3. Agregar, gota a gota, 10 ml. de la disolución de xanthidrol, agitando sin detenerse con un agitador de vidrio. Dejar reposar hasta que se forme el precipitado y agitar uno o dos minutos con el agitador, dejando después reposar durante hora y media.
10(a).4.4. Filtrar el contenido del vaso sobre un crisol con membrana filtrante de vidrio, previamente desecado a 130 °C y tarado. No debe emplearse más que un ligero vacío, lavando tres veces cada vez con 5 ml. de etanol, sin tratar de eliminar completamente el ácido acético. Desecar en la estufa a 130 °C durante una hora. La temperatura no debe pasar de 145 °C. Dejar enfriar en un desecador y pesar.
10(a).5. Cálculo.

m = peso del precipitado, en g.
v = volumen de la parte alícuota tomada, en ml.
10(a).6. Referencias.
1. «Método de la O. C. D. E.» Edición 1961, número 8.
10(b). NITROGENO UREICO (Método de p-dimetilaminobenzaldehído)
10(b).1. Principio.
La urea disuelta en agua se combina con el p-dimetilaminobenzaldehído en presencia de ácido clorhídrico, dando un compuesto soluble amarillo-verde. Medir la absorción de la luz para esta disolución a 420 nm. Si existen sustancias perturbadoras coloreadas y orgánicas, se las elimina por medio de carbón activado y de las disoluciones Carrez.
Es aplicable a todos los abonos que contengan una cantidad de urea correspondiente a un contenido de nitrógeno inferior al 3 por 100.
10(b).2. Material y aparatos.
10(b).2.1. Matraz aforado de 500 ml.
10(b).2.2. Agitador rotativo.
10(b).2.3. Pipeta de 5 ml.
10(b).2.4. Un espectrofotómetro o un absorciómetro que permita la absorción de la luz a 420 nm. con una célula de 2 a 3 centímetros de espesor.
10 (b).3. Reactivos.
10(b).3.1. Urea. Calidad para análisis.
10(b).3.2. Disolución de p-dimetilaminobenzaldehído: Disolver 1,6 g. de esta sustancia en 100 ml. de etanol de 96 por 100, agregando 10 ml. de ácido clorhídrico concentrado de densidad 1,19.
10(b).3.3. Carbón activado (officinal).
10(b).3.4. Disolución de Carrez I (acetato de cinc): Disolver en agua 23,8 g. de (C2H3O2)2 Zn. 3H2O, o bien, 22 g. del dihidrato (C2H3O2)2 Zn. 2H2O, agregando 3 g. de ácido acético y completando a 100 ml. con agua.
10(b).3.5. Disolución de Carrez II (ferrocianuro potásico): Disolver en agua 10,6 g. de Fe (CN)6K, diluyendo a 100 ml.
10(b).4. Procedimiento.
10(b).4.1. Pesar una muestra que no contenga más de 40 mg. de urea (con preferencia 1 g.) con la aproximación de 0,2 mg. Ponerla en un matraz aforado a 500 ml. con 400 a 450 ml. de agua. Si es necesario, agregar 1 g. de carbón activado, 5 ml. de disolución de Carrez I y 5 ml. de disolución de Carrez II. Agitar durante treinta minutos en un agitador rotativo. Enrasar, homogeneizar y filtrar.
10(b).4.2. Tomar con pipeta 5 ml. exactos del filtrado limpio e incoloro, poniéndolos en un tubo de ensayo limpio y seco, o si conviene mejor, en una célula tubular del absorciómetro. Agregar con una pipeta 5 ml. exactos de la disolución de p-dimetilaminobenzaldehído. Mezclar y mantener el tubo en baño María a 20 °C durante quince minutos.
10(b).4.3. Tratar exactamente de la misma forma partes alícuotas de 5 ml. de la disolución valorada de urea que contiene, respectivamente 0, 100, 200, 300, 400 µg. de urea en 5 ml. Medir la absorción de la luz (absorbancia) por estas disoluciones a 420 nm. comparándolas con la que no tiene urea.
10(b).5. Cálculo.
La comparación de las medidas de absorción permite calcular la cantidad de urea contenida en los 5 ml. de la disolución que se analiza. El porcentaje de urea en la muestra será:

El porcentaje de nitrógeno de la urea en la muestra se obtiene multiplicando este resultado por 0,46654.
10(b).6. Observaciones.
10(b).6.1. La disolución de p-dimetilaminobenzaldehído no se conserva más de dos semanas
10(b).6.2. Todas las medidas de absorción de la luz deben hacerse a la misma temperatura.
10(b).6.3. La presencia de grandes cantidades de hidrazina y de semicarbacida perturban la dosificación.
10(b).6.4. Las disoluciones utilizadas para las medidas de absorción de luz deben ser perfectamente límpidas.
10(b).6.5. El método se aplica a todos los abonos, gracias a la eliminación, por medio del carbón activado y de las disoluciones de Carrez, de las sustancias que perturban por su color o por otra causa.
10(b).7. Referencias.
1. «Métodos de la O. C. D. E.» Edición 1961, número 9.
10(c). NITROGENO UREICO (Método de la ureasa)
10(c).1. Principio.
Agregar ureasa neutra a la disolución neutra del fertilizante, de la que se han eliminado los fosfatos y los compuestos solubles de calcio, valorando el carbonato amónico producido y deduciendo de esta valoración el nitrógeno ureico.
Es aplicable a todos los fertilizantes que contienen urea.
10(c).2. Material y aparatos.
10(c).2.1. Vasos de 200 ml.
10(c).2.2. Matraz aforado de 500 ml.
10(c).2.3. Papel Whatman número 12.
10(c).2.4. Erlenmeyer de 250 ml.
10(c).2.5. Buretas de 25 ml..
10(c).3. Reactivos.
10(c).3.1. Disolución neutra de ureasa: Utilizar disolución de ureasa comercial al 1 por 100. Disolver 1 g. de ureasa en polvo de agua y enrasar a 100 ml. con agua. Tomar 10 ml. de la disolución, ponerlos en un erlenmeyer de 250 ml.-, diluir con 50 ml. de agua y añadir cuatro gofas de púrpura de metilo. Agregar ClH N/10 hasta color púrpura rojizo y después valorar en retorno con NaOH N/10 hasta color verde. De la diferencia en ml. se calcula la cantidad de ClH N/10 requerida para neutralizar los 90 ml. restantes (corrientemente, uros 2 6 3 ml. para los 100). Agregar esta cantidad de ácido y agitar bien.
Comprobar la actividad enzimática de la ureasa periódicamente.
10(c).3.2. ClH N/10.
10(c).3.3. NaOH N/10.
10(c).3.4. Disolución de púrpura de metilo (disolución alcohólica al 0,1 por 100).
10(c).3.5. ClH 2N.
10(c).3.6. Disolución saturada de Ba (OH)2.
10(c).3.7. Disolución de CO3Na2 al 10 por 100.
10(c).4. Procedimiento.
10(c).4.1. Pesar 10 ± 0,01 g. de muestra, colocarla en un papel de filtro Whatman número 12 de 15 cm. de diámetro y lavar por arrastre con unos 300 ml. de agua en un matraz aforado de 500 mililitros. Añadir de 75 a 100 ml. de disolución saturada de Ba (OH)2 para precipitar los fosfatos, dejar sedimentar y probar con unas gotas de Ba (OH)2 si la precipitación fue completa.
10(c).4.2. Añadir 20 ml. de disolución de carbonato sódico al 10 por 100 para precipitar el exceso de Ba y cualquier sal de Ca soluble. Dejar sedimentar y ensayar si la precipitación fue completa.
10(c).4.3. Enrasar a 500 ml., agitar, filtrar por papel Whatman número 12 y transferir una alícuota de 50 ml. (equivalente a 1 g. de la muestra) a un erlenmeyer de 250 ml. Añadir una o dos gotas de púrpura de metilo. Neutralizar con ClH 2N y añadir 2 ó 3 gotas en exceso. Neutralizar la disolución con sosa N/10 hasta el primer cambio de color del indicador.
10(c).4.4. Añadir 20 ml. de disolución neutra de ureasa. Tapar el matraz eon tapón de goma y dejar en reposo durante una hora a 20-25 °C. Enfriar el matraz en mezcla de hielo y agua y valorar inmediatamente con ClH N/10 hasta color púrpura completo. Luego añadir unos 5 ml. en exceso. Anotar el volumen total añadido y valorar por retomo' el ClH en exceso con NaOH N/10 hasta punto final neutro.
10(c).s. Cálculo.

P = peso de la muestra contenida en la alícuota, en g.
10(c).6. Observaciones.
10(c).6.1. Cuando se trate de disoluciones coloreadas, puede hacerse la valoración con pH-metro.
10(c).6.2. En lugar de tomar las cantidades indicadas en 10 (c).4.1 se puede pesar 5 g. de la muestra y utilizar un matraz de 1.000 ml. Así, al tomar una alícuota de 10 ml., aun cuando el abono sea muy rico en urea, son suficientes 20 ml. de disolución de ureasa para hidrolizarla.
10(c).7. Referencias.
1. «Métodos A.O.A.C.» Edición 1970, números 2.070 y 2.071.
11. BIURET (Método colorimétrico)
11.1. Principio.
El método está basado en la formación de un complejo entre el biuret y el sulfato de cobre en medio alcalino. Este complejo puede dosificarse midiendo el color por medio de un colorímetro o espectrofotómetro, comparando con patrones.
Es aplicable a la urea y a los fertilizantes que la contengan.
11.2. Material y aparatos.
11.2.1. Matraces de 100 ml.
11.2.2. Pipetas de 5 ml.
11.2.3. Colorímetro o espectrofotómetro.
11.3. Reactivos.
11.3.1. Disolución alcalina de tartrato: Disolver 40 g. de NaOH en 500 ml. de agua, enfriar, añadir 50 g. de ClH4O6 NaK, 4H2O y diluir hasta un litro. Dejar en reposo un día antes de usarse.
11.3.2. Disolución de sulfato de cobre: Disolver 15 g. de SO4Cu ⋅ 5H2O4H20 en agua libre de COa y diluir a un litro.
11.3.3. Disolución patrón de biuret (1 mg/ml.l: Disolver 100 mg. de biuret de calidad reactivo en agua libre de CO2 y diluir hasta 100 ml. Hay que prepararla la víspera porque se disuelve mal.
11.3.4. Resina de cambio iónico: Llenar una bureta de 50 ml. con una columna de 30 cm. de resma Amberlite IR 120 (H) sobre taco de lana de vidrio. Regenerar la columna después de cada uso, haciendo pasar 100 ml. de SO4H2 (1 - 9) o ClH (1 + 4) a través de la columna en una proporción aproximada de 5 ml. por minuto y luego lavar con agua hasta que el pH del efluente sea mayor que 6.
11.4. Procedimiento.
11.4.1. Preparación de la curva patrón: Transferir una serie de alícuotas de 2 a 50 ml. de disolución patrón de biuret a matraces de 100 ml.
11.4.2. Ajustar el volumen hasta unos 50.ml. con agua libre de CO2, añadir una gota de rojo de metilo y neutralizar con SO4H2 N/10 hasta color rosa. Añadir agitando 20 ml. de disolución alcalina de tartrato y luego 20 ml. de disolución de sulfato de cobre. Diluir hasta volumen, agitar sacudiendo diez segundos y colocar en baño de agua a 30° ± 5 °C. durante quince minutos. También preparar un ensayo en blanco.
Determinar la absorbancia de cada disolución por comparación con el ensayo en blanco a 555 nm. (también resulta satisfactorio un colorímetro con filtro de 500 a 570 nm.), con cubeta de 2 a 4 cm. Dibujar la curva patrón representando la absorbancia frente a los mg. de biuret de cada alícuota.
11.4.3. En urea: Agitar continuamente 2 a 5 g. de muestra en 100 ml. de agua, a unos 50 °C. durante treinta minutos. Filtrar y lavar en matraz de 250 ml., y enrasar. Pipetear una alícuota de 25 ml. a un matraz de 100 ml. y continuar como en 11.4.2.
11.4.4. En mezclas de fertilizantes: Agitar continuamente 10 a 20 g. de muestra en 150 ml. de agua, a unos 50 °C., durante treinta minutos. Filtrar y lavar en un matraz aforado de 250 ml. enrasado. Transferir una alícuota de 25 ml. a la columna de resina y ajustar el caudal a 4 ó 5 ml. por minuto. Recoger el eluido en un vaso de 100 ml. Cuando el nivel del líquido llega a la parte superior de la columna de resina, lavar con dos porciones de 25 ml. de agua. Al eluido y al líquido de lavado añadir dos gotas de rojo de metilo y luego NaOH N hasta color amarillo. Añadir SO4H2 N/10 hasta el momento justo en que la disolución vire a color rosa, transfiriéndolo a un matraz aforado de 100 ml., y enrasado con agua libre de CO2. Pipetear una alícuota de 50 ml. a un matraz aforado de 100 ml. y continuar como en 11.4.2.
11.5. Cálculo.
Determinar en la curva patrón, a partir de la absorbancia leída, los mg. de biuret contenidos en la alícuota. Los porcentajes de biuret correspondientes son:
11.5.1. En urea:

11.5.2. En mezclas fertilizantes:

11.6. Referencias.
1. «Métodos de análisis de la A. O. A. C.« Edición 1970, números 2.073, 2.074 y 2.075.
12. NITROGENO CIANAMIDICO
12.1. Principio.
Precipitar la cianamida como cianamida de plata y determinar el nitrógeno en el precipitado por el método Kjeldahl.
Aplicable a todos los abonos, incluidos los abonos compuestos, que contengan nitrógeno en cualquier forma.
12.2. Material y aparatos.
12.2.1. Matraces aforados de Stohmann de un litro.
12.2.2. El resto como 6(a).2.
12.3. Reactivos.
12.3.1. Acido acético glacial, calidad para análisis.
12.3.2. Amoníaco (10 por 100 en peso, o sea, 5,0 N, densidad 0,90).
12.3.3. Disolución de acetato de plata: Disolver 100 g. de acetato de plata en la disolución de amoníaco al 10 por 100 y completar a un litro con agua.
12.3.4. El resto como en 6(a).3.
12.4. Procedimiento.
12.4.1. Pesar de 4,9 a 5,1 g. de la muestra preparada para análisis, colocándolos en un matraz aforado de Stohmann de un litro. Agregar 400 ml. de agua y 15 ml. de ácido acético glacial. Agitar en un agitador rotativo durante una hora a 30 ó 40 vueltas por minuto. Filtrar en un matraz aforado de un litro. Lavar el residuo insoluble con un poco de agua y volverlo al matraz aforado de Stohmann. Agregar otros 400 ml. de agua y 15 ml. de ácido acético glacial agitando durante una hora. Filtrar sobre el mismo filtro que para la primera filtración, lavando el filtro y el residuo con un poco de agua. Agregar el filtrado y el agua de lavado al primer filtrado y completar a un litro.
El filtrado debe ser analizado inmediatamente después de su preparación.
12.4.2. Tomar con pipeta 50 ml. del filtrado poniéndolos en un vaso de 250 ml. y agregar amoníaco al 10 por 100 hasta reacción débilmente alcalina. Agregar 15 ml. de disolución caliente de acetato de plata para precipitar la cianamida de plata amarilla.
12.4.3. Filtrar y lavar el precipitado con agua destilada fría, hasta que esté completamente exento de amoníaco. Poner el filtro y el precipitado en un matraz Kjeldahl y determinar el nitrógeno como en el método Kjeldahl para muestras sin nitratos.
12.5. Cálculo.

A = ml., de NaOH N/10 consumidos en el análisis.
B = ml., de NaOH N/10 consumidos en un ensayo en blanco.
P = peso de la muestra contenida en la alícuota, en g.
12.6. Observaciones.
Como la cantidad de nitrógeno que se va a dosificar es débil, utilizar ácido sulfúrico N/10 y disolución de hidróxido sódico N/10 para dosificar el amoníaco destilado.
12.7. Referencias.
1. Métodos de la O. C. D. E.» Edición 1961, número 10.
13. NITROGENO INSOLUBLE EN AGUA
13.1. Principio.
Separar en un filtro la parte de abono que no se solubiliza en agua y determinar en ella el N total.
Aplicar cuando se pide separadamente el N soluble y el insoluble, como en los abonos y enmiendas orgánicos.
13.2. Material y aparatos.
Como en 6(a).2.
13.3. Reactivos.
13.3.1. Alcohol, etanol del 95 por 100.
13.3.2. El resto como en 6(a).3.
13.4. Procedimiento.
13.4.1. Pesar 2,5 g. de la muestra y colocar en un erlenmeyer de 50 ml. humedeciéndolo con alcohol. Añadir 20 ml. de agua y esperar quince minutos agitando de cuando en cuando.
Filtrar decantando por un filtro Whatman número 2, de 12 centímetros de diámetro.
Lavar cuatro o cinco veces con agua a la temperatura ambiente (20-25 °C) y decantar.
Finalmente, pasar todo el residuo al filtro.
13.4.2. Recoger el filtro con el residuo, introducirlo en un matraz Kjeldahl y determinar en ello el N total como en el método Kjeldahl para muestras sin nitratos.
13.5. Cálculo.
Como en 6(a).5.
13.6. Referencias.
1. «Método A O. A. C.», número 2 064.
14. NITROGENO ORGANICO
14.1. Principio.
Este método es muy semejante al de Robertson, salvo que al final se determinan también el N amoniacal y el N ureico para deducirlos, junto con el N nítrico, del N total.
14.2. Material y aparatos.
Como en 6(a).2.
14.3. Reactivos.
Como en 6(a).3, 7(b).3, 9.3 y 10(c).3, el método del formaldehído, el método de Robertson y el método de la ureasa.
14.4. Procedimiento.
14.4.1. Determinar el N total por el método Kjeldahl para muestras con nitratos o por el método comprensivo.
14.4.2. Separar y determinar el N insoluble en agua por el método correspondiente.
14.4.3. En sendas porciones del filtrado procedente del apartado anterior, determinar el N nítrico (por el método de Robertson), el N amoniacal (por el método del formaldehído) y el N ureico (por el método de la ureasa).
14.5. Cálculo.
N orgánico = I + T (I + N + A + U)
T = N total.
I = N insoluble
N = N nítrico.
A = N amoniacal.
U = N ureico.
14.6. Referencias.
1. Métodos de la A. O. A. C.». Edición 1970, números 2.051 y siguiente.
15. FOSFORO TOTAL
15.1. Principio.
Tratar el abono con un ácido fuerte para solubilizar el fosfórico y precipitar con ácido citromolíbdico y quinoleína, recogiendo y pesando el precipitado amarillo de fosfomolibdato de quinoleína, del que se deduce el contenido en P2O5.
Se puede precipitar con un reactivo único denominado quimociaco, que contiene el ácido molíbdico cítrico y la quinoleína.
Es aplicable a todos los abonos en que se precise determinar le que se denomina fosfórico total, tales como fosfatos de roca, escorias de desfosforación y abonos orgánicos.
15.2. Material y aparatos.
15.2.1. Erlenmeyer de 300 ml. y matraces aforados:
15.2.2. Vasos de 250 ml.
15.2.3. Pipetas de 10 ml.
15.2.4. Buretas de 25 ml.
15.2.5. Vasos filtrantes G-4 con poros de 5 a 15 micras de diámetro.
15.2.6. Trompa de vacío.
15.2.7. Papel de fibra de vidrio.
15.3. Reactivos.
15.3.1. NO3H concentrado.
15.3.2. ClH concentrado.
15.3.3. Nitrato sódico o potásico.
15.3.4. ClO4H de 70-72 por 100.
15.3.5. Bromato potásico al 0,5 por 100.
15.3.6. Reactivo ácido molíbdico-cítrico: Disolver con agitación 54 g. de anhídrido molibdico (MoO3) 100 por 100 y 12 g. de NaOH en 400 ml. de agua caliente y enfriar.
Disolver 60 g. de ácido cítrico en una mezcla de 140 ml. de ClH y 200 ml. de agua y enfriar. Añadir gradualmente la disolución molíbdica a la cítrica, agitando. Enfriar, filtrar y diluir a un litro. (La disolución puede ser verde o azul, el color se intensifica por exposición a la luz.) Si es necesario añadir gota a gota disolución de bromato potásico al 0,5 por 100 hasta que el color verde se vuelva pálido. Guardar en frasco de polietileno en la oscuridad.
13.3.7. Disolución de quinoleína: Disolver 50 ml. de quinoleína sintética en una mezcla de 60 ml. de ClH y 300 ml. de agua, agitando. Enfriar, diluir a un litro y filtrar. Guardar en frasco de polietileno.
13.3.8. Reactivo quimociaco: Disolver 70 g. de molibdato sódico dihidratado en 150 ml. de agua. Disolver 60 g. de ácido cítrico en una mezcla de 85 ml. de NO3H y 150 ml. de agua y enfriar. Añadir gradualmente la disolución molíbdica a la cítrico-nítrica, agitando.
Disolver 5 ml. de quinoleína sintética en una mezcla de 35 ml. de NO3H y 100 ml. de agua. Añadir gradualmente esta disolución a la molibdicocitriconítrica, mezclando y dejando en reposo veinticuatro horas. Filtrar, añadir 280 ml. de acetona, diluir a un litro de agua y mezclarlo. Guardar en frascos de polietileno.
15.4. Procedimiento.
15.4.1. Preparación de la disolución.
Tratar un gramo de la muestra como en (I), (II), (III) o (IV) según la clase de abono:
(I) Adecuado para muestras que contengan pequeñas cantidades de materia orgánica.
Tratar el gramo de muestra con 30 ml. de NO3H y 3 a 5 ml. de ClH y hervir hasta destruir la materia orgánica (unos treinta minutos para fertilizantes líquidos y suspensiones).
(II) Adecuado para fertilizantes que contengan mucho fosfato de hierro o aluminio o para escorias básicas. Disolver el gramo de muestra en 15 a 30 ml. de ClH y 3 a 10 ml. de NO3H.
(III) Generalmente aplicable a materiales o mezclas que contengan grandes cantidades de materia orgánica.
Añadir primero unos 5 ml. de NO3H y después 20 a 30 ml. de SO4H2 al gramo de muestra en un matraz de 200 ml. Dejar en digestión y calentar suavemente si es necesario hasta que pase la violencia de la reacción. Añadir 2 a 4 g. de nitrato sódico o potásico, hervir, y cuando la disolución esté casi incolora, añadir otra pequeña cantidad de nitrato. También se puede añadir el nitrato en pequeñas porciones de cuando en cuando.
Cuando la disolución esté decolorada, enfriar y añadir 150 mililitros de agua, hirviendo unos pocos minutos.
(IV) Adecuado para todos los fertilizantes.
Hervir suavemente durante treinta o cuarenta y cinco minutos con 20-30 ml. de NO3H en matraz adecuado (preferentemente Kjeldahl para muestras que tengan gran cantidad de materia orgánica) para oxidar toda la materia fácilmente oxidable.
Enfriar y añadir 10 a 20 ml. de ClO4H de 70-72 por 100. Hervir muy suavemente hasta que la disolución sea incolora o casi incolora y aparezcan en el matraz densos vapores blancos. No se debe hervir nunca a sequedad (muy peligroso).
(Con muestras que tengan gran cantidad dé materia orgánica, la temperatura debe elevarse hasta el punto de producción de vapor, aproximadamente 170 °C, durante un periodo de al menos una hora).
Enfriar ligeramente, añadir 50 ml. de agua y hervir unos minutos.
15.4.2. Enfriar la disolución procedente de (I), (II), (III) o (IV) y transferir a un matraz aforado de 250 ml. Enrasar, mezclar y filtrar a través de un filtro seco.
15.4.3. En un erlenmeyer de 500 ml., pipetear una alícuota del filtrado que no contenga más de 25 mg. de P2O5, diluir a unos 100 ml. con agua y continuar por una de las modalidades siguientes:
15.4.4. Método gravimétrico como fosfomolibdato de quinoleína.
Modalidad A.
Añadir 30 ml. de reactivo ácido cítrico-molíbdico y hervir suavemente durante tres minutos. (La disolución debe permanecer libre de precipitado durante este período.)
Retirar del calor y agitar suavemente. Añadir inmediatamente desde una bureta 10 ml. de disolución de quinoleína con agitación continua. (Añadir primero 3-4 ml. gota a gota y el resto en chorro continuo.)
15.4.5. Enfriar a temperatura ambiente, agitar cuidadosamente formado remolinos tres o cuatro veces durante el enfriamiento.
Filtrar por un gooch con papel de filtro de fibra de vidrio previamente desecado a 250 °C y tarado, lavar cinco veces con porciones de 25 ml. de agua.
Desecar el crisol con su contenido a 250 °C durante treinta minutos, enfriar en desecador y pesar como fosfomolibdato de quinoleína (C9H7N)3 H3 (PO4 ⋅ 12 MoO3). Efectuar paralelamente un ensayo en blanco.
15.4.0. Modalidad B.
Añadir 50 ml. del reactivo quimocíaco, cubrir con vidrio de reloj, colocar en placa caliente con vitrina bien ventilada y hervir durante un minuto.
Continuar como en 15.4.5.
15.5. Cálculo.
Tanto para la modalidad A como para la B:
P = peso de la muestra contenida en la alícuota, en g.
M = peso del precipitado de fosfomolibdato de quinoloína, en g.

15.6. Observaciones.
15.6.1. En lugar de gooch y papel de fibra de vidrio puede utilizarse un crisol filtrante de 5-15 mieras de diámetro de poros, tal como un G-4.
15.6.2. En vez de desecar el precipitado a 250 °C durante media hora, da el mismo resultado desecar a 150 °C durante una noche.
15.7. Referencias.
1. «Métodos oficiales A. O. A. C». Edición 1970, números 2.016 y 2.023 a.2.025.
2. Lotti, G., y Galoppini, C., Guida alie analisi Chimico agrarie, pág. 359. «Determinazione dell anidride fosfórica totale (método Officiale)». Bologna, 1967.
16. FOSFORO SOLUBLE EN AGUA
16.1. Principio.
Extraer el fósforo soluble en agua existente en la muestra y determinar su cantidad en la disolución acuosa según el método correspondiente al fósforo total (apartado 15.4.4 y siguientes).
Es aplicable a todos los abonos en que se exige por separado las riquezas garantizadas de anhídrido fosfórico soluble en agua y soluble en citrato amónico, tales como los superfosfatos.
16.2. Material y aparatos.
Como en 15.2.
16.3. Reactivos.
16.3.1. NO3H concentrado.
16.3.2. N03H diluido (1 + 1).
16.3.3. Reactivo quimocíaco. Como en 15.3.8.
16.4. Procedimiento.
16.4.1. Preparación de la disolución.
Colocar un gramo de muestra en un filtro de 6 cm. de diámetro y lavar con pequeñas porciones de agua hasta que el filtrado mida algo menos de 250 ml.
Dejar pasar cada porción a través del filtro antes de añadir más. Si el lavado no puede completarse al cabo de una hora, utilizar succión. En caso de que el filtrado esté turbio, añadir uno o dos ml. de NO3H concentrado. Enrasar a 250 ml. con agua y agitar (Disolución I).
16.4.2. En un erlenmeyer de 500 ml. pipetear una alícuota que no contenga más de 25 mg de P2O5 y diluir a 50 ml. si es necesario. Añadir 10 ml. de NO3H diluido (1 + 1), hirviendo durante diez minutos.
16.4.3. Método gravimétrico como fosfomolibdato de quinoleína. Como en 15.4.4, 15.4.5 y 15.4.6.
16.5. Cálculo.
Como en 15.5.
16.6. Observaciones.
Como en 15.6.
16.7. Referencias.
1. «Métodos Oficiales A. O. A. C.», edición 1970, números 2.032 y 2.025.
17. FOSFORO SOLUBLE EN AGUA Y EN CITRATO AMONICO NEUTRO (Asimilable)
17.1. Principio.
Después de separar el fósforo soluble en agua de la muestra, según el método de determinación del fósforo soluble en agua, someter el residuo a una extracción con disolución neutra (pH = 7,0) de citrato amónico. El fósforo asimilable puede determinarse en la disolución obtenida al reunir los extractos acuosos y de citrato amónico. También puede procederse a la determinación en los extractos separados. En ambos casos, proceder de acuerdo con el método de fósforo total (15.4.4 y siguientes).
Aplicable a aquellos abonos en que se exige el fósforo soluble en agua y en citrato conjuntamente, tales como los abonos compuestos y también para los superfosfatos.
17.2. Material y aparatos.
17.2.1. Un pH-metro.
17.2.2. Un aparato calentador agitador.
17.2.3. El resto como en 15.2.
17.3. Reactivos.
17.3.1. Citrato amónico neutro.
Debe tener un peso específico de 1,09 a 20 °C y un pH igual a 7,0, determinado electrométricamente.
Disolver 370 g. de ácido cítrico cristalizado en 1,5 litros da agua y casi neutralizar, añadiendo 345 ml. de hidróxido amónico (de 28 a 29 por 100 de NH3). Si la concentración de amoníaco es menor del 28 por 100, añadir mayor volumen y disolver el ácido cítrico en un volumen más pequeño de agua. Enfriar y comprobar el pH. Ajustar con hidróxido amónico (1 + 7) o con disolución de ácido cítrico a pH = 7.0. Si es preciso, diluir la disolución para que el peso específico sea 1,09 a 20 °C. El volumen será aproximadamente de dos litros.
Conservar en frascos herméticamente cerrados y comprobar el pH de cuando en cuando, reajustándolo si difiere de 7,0.
17.3.2. Reactivo quimocíaco. Como en 15.3.6.
17.4. Procedimiento.
17.4.1. Preparación de las disoluciones.
Después de retirar el P3O5 soluble en agua (disolución I) como en el método del fósforo soluble en agua, transferir el filtro y el residuo, en un tiempo no superior a una hora, a un matraz de 250 ml. que contenga 100 ml. de la disolución de citrato amónico neutro previamente calentada a 65 °C.
Cerrar el matraz herméticamente con tapón de goma compacto sacudir enérgicamente hasta que el papel se reduzca a pulpa y reducir la presión quitando momentáneamente el tapón.
Agitar constantemente el matraz tapado en un aparato que lo mantenga a 65 °C. La acción del aparato debe ser tal que la dispersión de la muestra en la disolución de citrato se mantenga continuamente y la superficie interna del matraz y el tapón se bañe constantemente en la disolución.
17.4.2. Exactamente una hora después de añadir el filtro y el residuo, quitar el matraz del aparato e inmediatamente filtrar el contenido por succión, tan rápidamente como sea posible, a través de papel Whatman número 5 o equivalente, utilizando un buchner o embudo corriente con cono de platino u otro material.
Lavar con agua a 65 °C hasta que el volumen del filtrado sea aproximadamente de 200 ml., dejando tiempo para que el drenaje sea completo antes de añadir más agua. Si el material es tal que produce filtrado turbio, lavar con disolución de nitrato amónico al 5 por 100. Enfriar y enrasar (disolución II).
Tomar una alícuota de la disolución I y otra de la disolución II, ponerlas en un erlenmeyer de 400 ml. (entre las dos alícuotas no deben tener más de 25 mg. de P2O5) y diluir a 50 ml. si es necesario.
17.4.3. Método gravimétrico como fosfomolibdato de quinolema. Como en 15.4.4, 15.4.5 y 15.4.6.
17.5. Cálculo.
Como en 15.5.
17. Observaciones.
Si no se dispone de un aparato agitador calentador puede sustituirse por un baño de agua a 65 °C, en el cual se introduce el matraz durante una hora, agitando de cuando en cuando.
El resto, como en 15.6.
17.7. Referencias.
1. «Métodos Oficiales A. O. A. C.». Edición 1970, números 2.032, 2.025, 2.037 y 2.038.
18. FOSFORO SOLUBLE EN CITRATO AMONICO ALCALINO (Petermann)
18.1. Principio.
Someter el abono directamente a la extracción por una disolución alcalina de citrato amónico (disolución de Petermann) y dosificar el fósforo disuelto como fosfomolibdato de quinoleína. Este método es muy semejante al del fósforo soluble en agua y citrato amónico neutro, pero se tiene en cuenta el mayor contenido de amoníaco en las observaciones que hace, ya que en aquél no se utiliza citrato alcalino.
Es aplicable al fosfato bicálcico.
18.2. Material y reactivos.
18.2.1. Embudo con llave.
18.2.2. Matraz Stohmann.
18.2.3. El resto, como en 15.2.
18.3. Reactivos.
18.3.1. Citrato amónico alcalino (disolución de Petermann):
Disolver en agua 173 g. de ácido cítrico (C6H8O7. H2O) y enfriando esta disolución agregarla a una cantidad suficiente de amoníaco (densidad = 0,91) para que la disolución final contenga 42,0 g. de nitrógeno por litro. Es preciso tomar precauciones para evitar una pérdida de amoníaco durante esta operación. La disolución de ácido cítrico debe agregarse por medio de un embudo con llave que cierre herméticamente el cuello del matraz que contiene la disolución amoniacal refrigerada. El aire desplazado atraviesa la disolución de ácido cítrico en el embudo con llave de modo que no hay pérdida de amoníaco. Enfriar a 15 °C y completar el volumen final con agua.
La densidad de la disolución obtenida debe estar comprendida entre 1,082 y 1,083. El contenido de nitrógeno debe ser de 42,0 g. por litro, es decir, que 25 ml. de una disolución al décimo (1 + 0) debe contener 0,1050 g. de nitrógeno.
18.3.2. Disolución de molibdato.
Pesar 54 g. de anhídrido molíbdico (MoO3), ponerlos en un vaso de 500 ml., agregar 200 ml. de agua y después, con agitación continua, 11 g. de hidróxido sódico sólido. Calentar el vaso hasta la disolución del anhídrido molíbdico.
Disolver 60 g. de ácido cítrico cristalizado en 250 ml. de agua en un vaso de un litro y agregar 140 ml. de ácido clorhídrico de densidad 1,16.
Verter la disolución molíbdica sobre la disolución cítrica, agitando vigorosamente sin detenerse. Enfriar, filtrar y enrasar a un litro con agua. Conservar en la oscuridad en frasco de polietileno bien cerrado.
18.3.3. Disolución de quinoleína.
Diluir 60 ml. de ClH concentrado, colocados en un vaso de un litro con 300 a 400 ml. de agua y calentando a 70 u 80 °C. Agregar 50 ml. de quinoleína sintética pura en chorro delgado, agitando constantemente.
Una vez disuelta la quinoleína, enfriar la disolución, diluir con agua, filtrar en matraz aforado y enrasar a un litro. Conservar en frasco de polietileno.
La quinoleína debe ser de la mayor pureza y exenta de sustancias reductores.
18.4. Procedimiento.
18.4.1. Preparación de la disolución.
Pesar un gramo de la muestra triturada y bien homogeneizada y ponerla en un mortero pequeño. Triturar y mezclar con porciones sucesivas de 5 ml. de la disolución de citrato amónico Petermann, agregados por medio de una bureta de 100 mililitros. Después de cada adición decantar el líquido sobrenadante a un matraz Stohmann aforado a 250 ml. y proseguir la operación hasta que toda la muestra tomada se reduzca a un estado de pasta lisa. Lavar el mortero con la disolución de Petermann y verter la disolución de lavado en el matraz aforado, no empleando como máximo más de 100 ml. de la disolución para la trituración y el lavado.
18.4.2. Agitar el matraz en un agitador rotativo durante tres horas a 30 ó 40 vueltas por minuto. Colocar el matraz en un baño maría a 40 °C. durante una hora, agitar por lo menos durante este tiempo con cuidado a mano cuatro veces. Enfriar, completar hasta la marca con agua, mezclar bien y filtrar por filtro con pliegues. El fósforo se dosifica en una parte alícuota conveniente del filtrado por el método de Perrin-Wilson.
18.4.3. Método de Perrin-Wilson.
Tomar una alícuota que no tenga más de 25 mg. de P2O5 y ponerla en un matraz de 250 ml.
Ver las observaciones relativas a las técnicas empleadas, en presencia de amoníaco, fosfatos complejos o sustancias reductores que figuran en el apartado 18.6.
18.4.4. Diluir con agua hasta un volumen aproximado de 110 ml. y agregar 25 ml. de disolución de molibdato, llevándolo a ebullición. Ponerlo en un baño maría (previamente calentado a ebullición) durante quince minutos. No debe formarse precipitado permanente hasta este momento.
18.4.5. Agregar con una bureta 12,5 ml. de disolución de quinoleína, los primeros ml. gota a gota y el resto en chorro delgado, haciendo girar constantemente el matraz de modo que se produzca un precipitado cuyas partículas sean del grosor máximo. Agitar el matraz durante tres minutos y enfriarlo, colocándolo al agua del grifo (20° ± 1 °C).
18.4.6. Filtrar por un crisol de vidrio o porcelana (diámetro de los poros, 5 a 15 mieras) previamente desecado a 250 °C y tarado. Lavar el precipitado con agua con el chorro del 'fraseó lavador, arrastrando sobre el crisol hasta las Ultimas trazas de precipitado contenido en el matraz. Desecar el crisol con el precipitado durante quince minutos a 250 °C, enfriar y pesar.
18.5. Cálculo.
Como en 15.5.
18.6. Observaciones.
18.6.1. Si la parte alícuota tomada contiene más de 100 miligramos de amoníaco proveniente del abono, agregar 1,25 g. de bicarbonato sódico y 25 ml. de agua, haciendo hervir suavemente y con precaución durante quince o dieciséis minutos. Enfriar, agregar lentamente y con cuidado 16 ml. de ClH 3N y hacer hervir suavemente durante unos minutos para expulsar la mayor parte del anhídrido carbónico.
18.6.2. Si la disolución en la cual se debe dosificar el fósforo ha sido preparada por medio de una disolución de citrato amónico neutro o alcalino, agregar, en lugar del bicarbonato sódico indicado en la observación anterior, 1 ml. de disolución de hidróxido sódico 3N por ml. de disolución de citrato amónico existente en la alícuota, así como 25 ml. de agua, haciendo hervir suavemente durante quince minutos.
Enfriar y agregar tantos ml. de ClH 3N como se ha agregado de la disolución de sosa 3N.
18.6.3. Si la disolución en la cual se debe dosificar el fósforo contiene metafosfatos y/o fosfatos polimerizados, agregar a la parte alícuota (después del tratamiento para eliminar el amoníaco. si fuera preciso) un ml. de ClH concentrado, y hervir durante quince minutos.
18.6.4. Si la disolución en la cual se debe dosificar el fósforo proviene de una muestra de escorias o si por cualquier otra razón se la considera como susceptible de contener sustancias reductoras, utilizar la técnica indicada en la observación anterior y agregar además 2 ml. de una disolución de clorato potásico al 1,25 por 100 antes de hacer hervir durante quince minutos.
En el caso en que se utilice una o varias de las técnicas mencionadas en estas observaciones, proseguir como en el apartado 18.4.4.
18.7. Referencias.
1. -Métodos O. C. D. E.». Edición 1961, números 16 y 18.
19. FOSFORO SOLUBLE EN ACIDO CITRICO AL 2 POR 100
19.1. Principio.
Someter el abono a una extracción por la disolución de ácido cítrico al 2 por 100, destruyendo las sustancias reductoras con ClH y clorato potásico. La precipitación se hace al estado de fosfomolibdato de quinoleína por el método de Perrin-Wilson, teniendo en cuenta sus observaciones.
Es aplicable a todos los abonos en los que se garantice un contenido de P2O5 soluble al ácido cítrico al 2 por 100, como en las escorias de desfosforación.
19.2. Material y aparatos.
19.2.1. Frascos Stohmann.
19.2.2. Agitador rotativo.
19.2.3. El resto, como en 15.2.
19.3. Reactivos.
19.3.1. Disolución de ácido cítrico.
Disolver en agua 20 g. de ácido cítrico cristalizado y enrasar a un litro con agua. Comprobar la concentración en ácido cítrico de esta disolución, valorando 10 ml. de ella con NaOH N/10 en presencia de fenolftaleína. Si la disolución está bien hecha se deben gastar 28,55 ml.
19.3.2. Disolución de molibdato.
Como en 18.3.2.
19.3.3. Disolución de quinoleína.
Como en 18.3.3.
19.4. Procedimiento.
19.4.1. Preparación de la disolución.
Pesar de 4,9 a 5,1 g. de muestra con la aproximación de 1 miligramo. Colocarlos en un matraz Stohmann de un litro y agregar con agitación a mano 500 ml. de la disolución de ácido cítrico. Agitar el matraz en un agitador rotativo durante treinta minutos a 30 ó 40 vueltas por minuto. Durante la agitación, mantener la temperatura ambiente a 20° ± 1 °C.
Filtrar inmediatamente sobre un filtro rápido y tomar una alícuota conveniente para la dosificación del fósforo en el filtrado por el método de Perrin-Wilson.
19.4.2. Método de Perrin-Wilson.
Ver el método de fósforo soluble en citrato amónico alcalino.
19.5. Cálculo.
Como en 15.5.
19.6. Observaciones.
19.6.1. Si no se dispone de matraces Stohmann se pueden utilizar matraces o frascos corrientes aforados..
19.6.2. La precipitación del fosfórico debe hacerse sobre el filtrado recientemente obtenido.
19.7. Referencias.
1. «Métodos O. C. D. E.». Edición 1961, número 14.
DETERMINACION DEL POTASIO
20(a). POTASION SOLUBLE EN AGUA (Método gravimétrico)
20(a).1. Principio.
Precipitar el potasio en disolución débilmente alcalina por el tetrafenilborato de sodio B (C6H5)4 Na, desecar y pesar el precipitado de tetrafenilborato de potasio.
Previamente hacer hervir para eliminar la mayor parte del amoníaco que pudiera eventualmente existir y el resto es transformado en hexametileno tetramina por adición de formol. La adición de E. D. T. A. impide que ciertos metales puedan perturbar la dosificación.
Es aplicable a todos los abonos potásicos solubles en agua.
20(a).2. Material y aparatos.
20(a).2.1. Vasos de 250 ml. y 400 ml.
20(a).2.2. Matraces aforados de 500 ml. y 1.000 ml.
20(a).2.3. Pipetas graduadas de dos trazos y de diversas dimensiones.
20(a).2.4. Crisol de vidrio o porcelana filtrante, cuyos poros tengan un diámetro de 10 a 20 mieras.
20 (a).3. Reactivos.
20(a).3.1. Hidróxido de sodio, disolución 10N.
20(a).3.2. E. D. T. A.: Disolver 4 g. de dihidrato de etileno diamino tetracetato disódico en agua y completar a 100 ml.
20(a).3.3. T. P. B. S.: Disolver 32,5 g. de tetrafenilborato de sodio en 480 ml. de agua, añadir 2 ml. de disolución de hidróxido sódico 10N y 20 ml. de disolución de cloruro de magnesio (100 gramos de Cl2Mg. 6H2O por litro), agitar con un agitador magnético durante quince minutos y filtrar a través de un filtro duro de poros finos.
20(a).3.4. Formol, disolución al 25 por 100.
20(a).3.5. Fenolftaleína: Disolver 5 g. de fenolftaleína en etanol al 90 por 100 y completar a un litro.
20(a).3.6. Líquido de lavado: Diluir 2 ml. de reactivo 20(a).3.3 hasta un volumen de 10 ml. con agua.
20(a).4. Procedimiento.
20(a):4.1. Pesar una muestra de 5 g. con la aproximación de 1 mg„ añadir 400 ml. de agua y hervir durante treinta minutos. Enfriar y completar a 500 ml. en un matraz aforado. Mezclar y filtrar tirando los primeros 50 ml. del filtrado.
20(a).4.2. Tomar con la pipeta una parte alícuota [ver 20(a).6.1] que no contenga más de 60 mg. de K2O y de un volumen que no sobrepase de 50 ml. Colocar en un vaso de 250 ml. y completar hasta un volumen de 50 ml. en caso necesario.
20(a).4.3. Añadir 10 ml. de disolución E. D. T. A., algunas gotas de fenolftaleína y gota a gota de disolución de hidróxido de sodio hasta coloración roja. Añadir unas gotas de disolución de hidróxido de sodio en exceso. Si se trata de un abono compuesto conteniendo compuestos amoniacales, procurar hervir suavemente y con precaución durante un cuarto de hora para eliminar la mayor parte del amoníaco y después añadir agua hasta llegar al volumen inicial.
20(a).4.4. Calentar hasta ebullición y añadir 10 ml. de formol, y además algunas gotas de disolución de hidróxido de sodio hasta coloración roja bien marcada. Cubrir el vaso con un vidrio de reloj y calentar durante quince minutos al baño maría.
20(a).4.5. Añadir gota a gota y agitando 10 ml. de disolución de T. P. B. S. Continuar agitando un minuto y enfriar después rápidamente.
20(a).4.6. Dejar reposar diez minutos y filtrar sobre un crisol previamente seco y pesado cuyo diámetro de poros esté comprendido entre 5 y 15 micras. Lavar tres veces con el líquido de lavado y una vez con 5 ml. de agua.
20(a).4.7. Secar el crisol y su contenido durante una hora y media a 120 °C. Dejar enfriar en un desecador y pesar.
20(a).5. Cálculo.
Un gramo de precipitado seco equivale a 0,1091 g. de K o 0,1314 g. de K2O. Luego,

m = peso del precipitado, en g.
v = volumen de la alícuota tomada, en ml.
20(a).6. Observaciones.
20(a).6.1. Si el filtrado obtenido después de la preparación de la disolución estuviera turbio o de color oscuro o fuese susceptible de contener ácido silícico disuelto, colocar una parte alícuota que contenga como máximo 120 mg. de K2O en un matraz aforado de 100 ml., añadir agua de bromo y hacer hervir hasta que el bromo en exceso se haya eliminado. Enfriar, completar a 100 ml. con agua y filtrar. Valorar el potasio en una parte alícuota conveniente del filtrado, según la técnica indicada más arriba.
20(a).6.2. Si la pureza del precipitado es dudosa, por ejemplo, si no estuviese de un blanco puro, lavar con acetona después de desecado y pesado. El tetrafenilborato de potasio se disuelve fácilmente en la acetona. Secar, enfriar y pesar de nuevo el crisol. La diferencia representa el peso del tetrafenilborato de potasio.
20(a).7. Referencias.
1. «Método O. C. D. E.». Edición 1961, número 11.
20(b). POTASIO SOLUBLE EN AGUA (Método volumétrico)
20(b).1. Principio.
Precipitar el potasio de una disolución de la muestra (previamente tratada para eliminar posibles interferencias) con un exceso de disolución normalizada de tetrafenilboro sódico. Después de separar el precipitado de sal potásica, valorar el exceso de reactivo, con una disolución patrón de una sal de amonio cuaternario, usando como indicador amarillo de titanio (amarillo Clayton).
Se aplica a todos los abonos potásicos solubles en agua.
20(b).2. Material y aparatos.
20(b).2.1. Matraces aforados.
20(b).2.2. Vasos de 250 ml.
20(b).2.3. Microbureta de 10 ml.
20(b).3. Reactivos.
20(b).3.1. Disolución de hidróxido sódico al 20 por 100; Disolver 20 g. de NaOH en 100 ml. de H2O.
20(b).3.2. Disolución de formaldehído al 37 por 100.
20(b).3.3. Disolución de tetrafenilboro de sodio (TPBS), aproximadamente al 1,2 por 100. Disolver 12 g. de B (C6H5)4Na en aproximadamente 800 ml. de H2O. Añadir 20-25 g. de Al(OH)3, agitar cinco minutos y filtrar (papel Whatman numero 42 o equivalente) en un matraz de 1 litro. Lavar el vaso ligeramente con H2O y añadir al filtro. Recoger el filtrado entero, añadir 2 ml. de NaOH al 20 por 100, diluir hasta volumen con H2O y mezclar. Dejar en reposo cuarenta y ocho horas y normalizar. Ajustar de forma que 1 ml. de TPBS sea igual al 1 por 100 de K2O. Guardar a temperatura ambiente.
20(b).3.4. Disolución de cloruro amónico cuaternario, aproximadamente al 0,025 por 100: Diluir 50 ml. de cloruro de zefirán (disponible en farmacias como cloruros de alquildimetilbenzil amonio) al 12,8 por 100 hasta 1 litro con H2O, mezclar y normalizar. El bromuro de cetiltrimetilámonio puede sustituir al cloruro de zefirán. Si se utiliza otra concentración, ajustar el volumen.
20(b).3.5. Amarillo de titanio (Clayton Yelow; índice de color número 19.540) al 0,04 por 100: Disolver 40 mg. en 100 ml. de H2O.
20 (b).4. Procedimiento.
Normalización de las disoluciones.
20(b).4.1. Cloruro de zefirán: Añadir a 1 ml. de disolución de TPBS, en erlenmeyer de 125 ml., 20-25 ml. de H2O, 1 ml. de NaOH al 20 por 100, 2.5 ml. de HCHO, 1.5 ml. de C2O4 (NH4)2 al 4 por 100 y 0-8 gotas de indicador. Valorar hasta punto final rosa con disolución de cloruro de zefirán utilizando una bureta semimicro de 10 ml. Ajustar la disolución de cloruro de zefirán de forma que 2 ml. sea igual a 1 ml. de disolución TPBS.
20(b).4.2. Disolución de tetrafenilboro de sodio: Disolver 2,5 g. de PO4KH2 en H2O en matraz aforado de 250 ml., añadir 50 ml. de C2O4 (NH4)2 disolución al 4 por 100, diluir hasta volumen con H2O y se mezcla (no es necesario llevarla a ebullición). Transferir una alícuota de 15 ml. (51,92 mg. de K2O igual a 43,10 mg. de K) a matraz aforado de 100 ml., añadir 2 ml. de NaOH al 20 por 100, 5 ml. de HCHO y 43 ml. de reactivo TPBS. Diluir hasta volumen con H2O, mezclar por completo, dejar en reposo cinco-diez minutos y pasar a través del filtro seco. Transferir una alícuota de 50 ml. del filtrado a un erlenmeyer. de 125 ml., añadir 0-8 gotas de indicador y valorar el exceso de reactivo con disolución de zefirán. Calcular el título en la forma siguiente:
F = 34,61/ (43 ml. ml. de zefirán) = % K2O/ml. de reactivo TPBS.
Ei factor F se aplica a todos los fertilizantes, si se diluyen 2,5 g. de muestra hasta 250 ml. y se toma una alícuota de 15 ml. para análisis. Si los resultados van a expresarse como K en vez de como K2O, sustituir 34,61 por 28,73 en el cálculo de F.
20(b).4.3. Introducir una muestra de 2,5 g. (1,25 g. si K2O es superior al 50 por 100) en un matraz aforado de 250 ml., añadir 50 ml. de C204 (NH4)2 al 4 por 100 y 125 ml. de H2O y hervir durante treinta minutos (si hay presente materia orgánica, añadir 2 g. de carbón activado libre de K antes de la ebullición). Enfriar, diluir hasta volumen con H2O, mezclar y filtrar por filtro, seco o dejar en reposo hasta que quede clara. Transferir una alícuota de 15 ml. de disolución de muestra a un matraz aforado de 100 ml. y añadir 2 ml. de NaOH al 20 por 100 y 5 ml. de HCHO. Añadir 1 ml. de la disolución patrón TPBS por cada 1 por 100 (1/2 si se tomaron 1,25 g. de muestra) de K2O, que se supone contiene la muestra, más 8 ml. en exceso para asegurar la precipitación completa. Diluir hasta volumen con H2O, mezclar por completo, dejar en reposo cinco-diez minutos y filtrar por filtro seco (Whatman número 12 o equivalente). Transferir 50 ml. de filtrado a erlenmeyer de 125 ml., añadir seis-ocho gotas de indicador y valorar el exceso de reactivo con disolución patrón de zefirán.
20(b).5. Cálculo.
Porcentaje de K2O en la muestra igual a (ml. de TPBS añadidos menos ml. de zefirán) por F; donde F es igual a porcentaje de K2O/ml. de reactivo TPBS (multiplicar por dos si se ha utilizado 1,25 g. de muestra).
20(b).6. Referencias.
1. «Métodos de análisis A. O. A.C.», número 2.090.
20(c). POTASIO SOLUBLE EN AGUA (Método fotométrico de llama)
20(c).1. Principio.
Se funda en la comparación de la medida de intensidades de emisión de átomos excitados en una llama, de una disolución problema desconocida, y de una disolución patrón de concentración conocida.
Para evitar interferencias, eliminar los metales pesados por precipitación con oxalato amónico y los aniones interferentes por retención mediante resinas cambiadores.
Se aplica a todos los abonos potásicos solubles en agua.
20(c).2. Material y aparatos.
20(c).2.1. Matraces aforados de 250 ml. y 500 ml.
20(c).2.2. Pipetas de distintos tamaños.
20(c).2.3. Papel milimetrado.
20(c).2.4. Columna de cambio iónico: Se hace de tubo de vidrio de pared normalizada y 30,5 cm. de longitud y 2,5 cm. de diámetro exterior; cerrar un extremo con tapón de goma número 4, con un orifio a través del cual se inserta una llave de dos pasos o un tubo de vidrio conectado a un tubo de goma y al empalme del compresor. No dejar que el tubo de la llave sobresalga por encima del tapón. Elegir un tapón suficientemente grande para que no quede espacio entre el extremo del tapón y la pared de la columna. También puede utilizarse tuvo de vidrio de cromatografía de 30,5 cm. y 1,9 cm. de diámetro interno, con llave o válvula en el fondo para controlar el caudal de líquido.
Colocar un taco de lana de vidrio en el fondo de la columna, cerrar la válvula y añadir H2O hasta 10 cm. de altura. Transferir una porción de resina a un vaso de 200 ml. y suspender en H2O. Transferir la suspensión a la columna y ajustar lá altura de la resina compacta a 20 cm. Drenar el exceso de H2O hasta que queda a 2.5 cm. de la parte superior. Regenerar la resina después de que se hayan pasado 10 alícuotas sucesivas, excepto con amberlita IR-4B, con la que pueden utilizarse 20 alícuotas. Para el Na, regenerar después de hacer pasar cinco alícuotas.
20(c).2.5. Preparación de la resina: Colocar 450 g. de resina en vaso de cuatro litros y añadir dos litros de NaOH al 5 por 100. Agitar treinta minutos con agitador eléctrico. Dejar sedimentar la resina y decantar la disolución de NaOH. Repetir el tratamiento con NaOH al 5 por 100 dos veces más y decantar la disolución de NaOH después del tratamiento final. Añadir dos litros de H2O a la resina, agitar unos minutos, dejar la resina sedimentar y decantar el H2O de lavado. Repetir trescuatro veces. La resina quedará ahora en la forma de base libre. Regenerar a la forma NO3 tratando tres veces con NO3H al 5 por 100 en la misma forma que como con la disolución de NaOH, Lavar la resina con H2O hasta que el agua de lavado alcance pH 2 o superior mediante el lavado por retroceso en la columna o agitando y decantando en un vaso grande. Guardar la resina cubierta de H2O en frasco tapado.
20(c).2.6. Fotómetro de llama.
20(c).3. Reactivos.
20(c).3.1. Disolución de oxalato amónico: Disolver 40 g. de C2O4 (NH4)2 en un litro de H2O.
20(c).3.2. Indicador rojo de metilo: Disolver 0,2 g. de rojo de metilo en 100 ml. de alcohol.
20(c).3.3. Acido nítrico diluido (1 + 10).
20(c).3.4. Utilizar una resina de cambio aniónico de basicidad intermedia o débil, tal como amberlita IR-49, duolita A-7 o duolita A-41, de-acidita o permuata S o equivalente.
20(c).3.5. Nitrato potásico o cloruro potásico: Recristalizar dos veces a partir de H2O la sal de grado reactivo y desecar cinco horas a 105 °C.
20(c).4. Procedimiento.
20(c).4.1. Preparación de la disolución problema.
(A) Mezclas de fertilizantes y sulfato potásico magnésico. Pesar 1,5058 g. de muestra en matraz aforado de 250 ml. (utilizar matraz de 50 ml. si la muestra contiene más del 30 por 100 de K2O), añadir 125 ml. de H2O y 50 ml. de disolución C2O4 (NH4)2 y hervir treinta minutos. Enfriar, diluir hasta volumen, mezclar y pasar por filtro seco.
20(c).4.2. (B). Sulfato y cloruro potásico: Disolver 1,5058 g. en H2O y diluir hasta 500 ml.
20(c).4.3. Preparación de la curva patrón: Disolver 1,2931 g. de NO3K (o 0,9535 g. de ClK) en H2O y diluir hasta 500 ml. (1.000 p.p.m. de K). Preparar las disoluciones patrón por dilución cubriendo los límites 0-80 p.p.m. de K a intervalos no superiores a 10 p.p.m. y añadiendo la cantidad adecuada de NO3Li si va a utilizarse un instrumento con patrón interno. Preparar la curva patrón de emisión frente a concentración ajustando el instrumento de forma que 50 p.p.m. de K de lecturas próximas al centro de la escala. Atomizar porciones de disoluciones patrón hasta que las lecturas para las series sean reproducibles.
20(c).4.4. Determinación.
(A) Mezclas de fertilizantes, sulfato potásico y sulfato potásico magnésico. Transferir una alícuota de 10 ml. de disolución de la muestra a un vaso de 250 ml. Añadir una gota de rojo de metilo y neutralizar con NO3H (1 + 10). Ajustar el nivel de H2O en la columna hasta la parte superior de la resina y transferir cuantitativamente la alícuota a la columna. Abrir la llave para obtener una proporción de flujo de dos gotas/segundo, recoger el efluente en matraz aforado de 250 ml. Lavar la alícuota dentro de la resina con dos-tres pequeñas porciones de H2O. Recoger 50-75 ml. de efluente, luego abrir la llave y recoger 100 ml. adicionales vertiendo H2O en la columna, procurando que el nivel de H2O no quede por debajo de la parte superior de la columna de resina. Diluir hasta volumen y mezclar (si se utiliza instrumento con patrón interno, se necesita añadir la cantidad precisa de NO3Li antes de diluir a volumen).
20(c).4.5. Atomizar porciones de muestras varias veces para obtener lecturas medias seguras para cada disolución. Determinar las p.p.m. de K a partir de la curva patrón. (La temperatura de las disoluciones patrón y de la muestra no deben diferir en más de 2 °C).
20(c).4.6. (B). Cloruro potásico. Proceder como en (A), pero omitir la neutralización y el tratamiento con la resina.
20(c).4.7. Prueba de comportamiento del instrumento y el procedimiento.
Pasar 1,5058 g. de ftalato ácido de potasio (patrón primario) y transferir a un matraz aforado de 250 ml. Añadir 0,5 g. de PO4H (NH4)2 y proceder como en 20(c).4.1, empezando en «añadir 125 ml. de H2O...» % K2O, calculado debe ser igual a 23,0.
20(c).5. Cálculo.
Cuando el % de K2O es de 0 a 30, el % K2O = p.p.m. K/2.
Cuando el % de K2O es mayor de 30, el % K2O = p.p.m. K/l.
20 (c).6., Observaciones.
Las sales de 20(c).3.5 no es necesario recristalizarlas si se dispone de una pureza de grados espectrográficos.
20(c).7. Referencias.
1. «Métodos de análisis», A. O. A. C. Edición de 1970, número 2.084.
21. POTASIO TOTAL
21.1. Principio.
Consiste en solubilizar el potasio insoluble en agua, pero que puede ser útil como fertilizante, y determinar el total a continuación por el método gravimétrico u otro cualquiera.
Aplicable a los abonos orgánicos que no pueden ser analizados por los métodos anteriores.
21.2. Material y aparatos.
Como en 20 (a).2.
21.3. Reactivos.
21.3.1. ClH.
21.3.2. NO3H.
21.3.3. El resto, como en 20 (a).3.
21.4. Procedimiento.
21.4.1. Pesar 10 g. del problema y calcinar en una cápsula de platino lentamente, removiendo el contenido y no elevando mucho la temperatura. Obtenida la calcinación, agregar a la cápsula agua caliente, decantando cuidadosamente el contenido a un matraz aforado de 250 ml.
21.4.2. Volver a agregar a la cápsula agua acidulada con ClH, calentando con cuidado para que no haya proyecciones, lavar con agua caliente y agregar cinco gotas de NO3H, arrastrando todo el contenido de la cápsula al matraz en el que debe hervir el contenido durante unos diez minutos. Dejar enfriar a la temperatura ambiente, enrasar y agitar.
21.4.3. Filtrar, y del filtrado tomar 50 ml. que equivalen a 2 g. de problema, y evaporar en cápsula de platino hasta sequedad, calcinando ligeramente.
El residuo se trata por agua caliente y filtra, lavando escrupulosamente la cápsula y el filtro.
21.4.4. En el filtrado, determinar el potasio como en el método gravimétrico del potasio soluble en agua.
21.5. Cálculo.
Como en 20 (a).5.
21.6. Observaciones.
En lugar del método gravimétrico puede emplearse otro de los descritos para el potasio soluble en agua.
21.7. Referencias.
1. «Análisis de abonos», Ministerio de Agricultura, 1953. Madrid.
| Abreviaturas, símbolos, definiciones y notas | |
|---|---|
| atm. | atmósfera |
| Be. | Baumé |
| C. | centígrado |
| cm. | centímetro |
| d. | densidad |
| f. c. r. | fuerza centrífuga relativa |
| g. | gramo |
| h. | hora |
| Kg. | kilogramo |
| l. | litro |
| M. | molar |
| m. | metro |
| meq. | miliequivalente |
| mg. | miligramo |
| min. | minuto |
| ml. | mililitro |
| mm. | milímetro |
| nm. | nanómetro |
| N. | normal |
| P.e. | punto de ebullición |
| p.f. | punto de fusión |
| PH. | logaritmo cambiado de signo de la concentración de iones hidrógeno |
| p.p.m. | partes por millón |
| p/p. | peso a peso |
| p/v. | peso a volumen |
| Rf. | relación entre las distancias recorridas por el disolvente y por la sustancia en una separación cromatográfica |
| r.p.m. | revoluciones por minuto s. segundo |
| Tm. | tonelada métrica |
| v/v. | volumen a volumen |
| ° | grado |
| % | por ciento |
| µ | micra |
| µg | microgramo |
| µl | microlitro |
| Σ | suma de |
| / | dividido por |
| X | multiplicado por < menor que |
| > | mayor que |
| <> | equivalente a |
| ≈ | aproximado a |
Absorbancia: Logaritmo cambiado de signo de la relación de las transmitancias de la muestra y del material de referencia o patrón.
Estas unidades corresponden a las declaradas de uso obligatorio por la Ley 88/1967, de 8 de noviembre, y Decreto 1256/ 1974, de 25 de abril.
State Agency Official State Gazette
Avda. de Manoteras, 54 - 28050 Madrid




