Agencia Estatal Boletín Oficial del Estado






Excelentísimos señores:
Por Órdenes de 30 de noviembre de 1976 («Boletín Oficial del Estado» de 4 de enero de 1977) y de 31 de enero de 1977 («Boletín Oficial del Estado» de 14 de julio) se establecieron diversos métodos oficiales de análisis, contemplando en el apartado segundo la posibilidad de su ampliación a medida que los correspondientes grupos de trabajo avancen en el estudio de nuevos métodos. Por otra parte, el continuo progreso de las técnicas de análisis aconsejan la revisión periódica de estos métodos modificándolos, completándolos o sustituyéndolos.
En consecuencia, a propuesta de los Ministros de Defensa, de Hacienda, de Administración Territorial, de Sanidad y Seguridad Social, de Industria y Energía, de Comercio y Turismo y de Agricultura, esta Presidencia del Gobierno dispone:
Se aprueban como oficiales los métodos de análisis de aceites y grasas, productos cárnicos, cereales y derivados, fertilizantes, productos fitosanitarios, productos lácteos, piensos, aguas y productos derivados de la uva, que se citan respectivamente en los anejos del I al IX.
Cuando no existan métodos oficiales para determinados análisis y hasta que sean estudiados por el grupo de trabajo correspondiente, podrán ser utilizados los adoptados por Organismos nacionales o internacionales de reconocida solvencia.
Quedan derogadas las disposiciones de igual o inferior rango que se opongan a la presente Orden.
La presente disposición entrará en vigor a los treinta días de su publicación en el «Boletín Oficial del Estado».
Lo que comunico a VV.EE.
Madrid, 31 de julio de 1979.
PÉREZ-LLORCA Y RODRIGO
Métodos de análisis de aceites y grasas
10(b). ÍNDICE DE ACIDEZ
(Método potenciométrico)
10 (b).1 Principio.
La determinación de la acidez libre se efectuará por volumetría, utilizando indicador potenciométrico, siendo éste el método aplicable cuando la valoración intensa de la solución o su turbidez dificultan la apreciación del viraje del indicador coloreado.
10(b).2 Material y aparatos.
10(b).2.1 Equipo de valoración potenciométrica equipado con electrodos de vidrio/calomelano o de wolframio/calomelano.
10(b).2.2 Agitador magnético.
10(b).2.3 Bureta de 25 ml, dividida en décimas de ml.
10(b).2.4 Matraz aforado de 1.000 ml.
10(b).2.5 Probeta graduada de 50 ml.
10(b).2.6 Vasos de precipitado de 150 ml, forma alta.
10(b).3 Reactivos.
10(b).3.1 Isobutil-metil-cetona  = 0,8; p. e. = 114-117 °C.
= 0,8; p. e. = 114-117 °C.
10(b).3.2 lsopropanol.
10(b).3.3 Hidróxido potásico.
10(b).3.4 Ácido benzoico.
10(b).3.5 Disolución 0,1 N de hidróxido potásico en isopropanol.
Pesar 7 g de hidróxido potásico, en lentejas, e introducirlas en un matraz aforado de un litro. Adicionar alcohol isopropílico, agitar con agitador magnético hasta conseguir una disolución completa y enrasar. Esta disolución se valora con ácido benzoico.
10(b).4 Procedimiento.
10(b).4.1 Preparación de la muestra.
La muestra deberá estar seca y libre de materias extrañas en suspensión. En caso contrario, antes de proceder a la pesada, deberá decantarse el agua si hubiese lugar a ello; y, en todo caso, filtrar con papel de filtro, efectuándose esta operación a una temperatura ligeramente superior a la del punto de fusión de la grasa.
Las grasas que contengan ácidos grasos volátiles no podrán calentarse, debido al riesgo de volatilización de ácidos libres. En estos casos, se procederá a la determinación de la acidez directamente, refiriéndose a muestra seca y exenta de impurezas insolubles, basándose en las determinaciones realizadas sobre muestras independientes.
10(b).4.2 Determinación.
Pesar de 5 a 10 g de materia grasa en un vaso de 150 ml y agregar 50 ml del reactivo 10(b).3.1. A continuación introducir los electrodos y proceder a la valoración con la disolución de hidróxido potásico.
10(b).5 Cálculos.
Calcular el índice de acidez aplicando la siguiente fórmula:

A = volumen, en ml, consumidos en la valoración.
N = normalidad de la disolución de hidróxido potásico.
P = peso, en g, de la muestra.
10(b).6 Observaciones.
Cuando se utilicen electrodos simples la unión entre la disolución saturada de cloruro potásico y la disolución de medida es conveniente hacerla a través de una espiga de porcelana porosa de unos 3 cm de longitud o por cualquier otro sistema que impida una difusión apreciable entre ambas disoluciones durante el tiempo que dura la valoración.
10(b).7 Referencias.
1. Instituto Nacional de Racionalización y Normalización del Trabajo. Una Norma Española 55.063.
11(b). ÍNDICE DE SAPONIFICACIÓN
(Método potenciométrico)
11(b).1 Principio.
Se denomina índice de saponificación el peso en mg de hidróxido potásico necesario para saponificar 1 g de materia grasa.
La determinación se realiza saponificando la muestra con una disolución previamente valorada de hidróxido potásico, determinando por volumetría el exceso de hidróxido potásico, utilizando indicador potenciométrico. Este método debe aplicarse cuando la coloración intensa de la solución o turbidez dificulten la apreciación del viraje del indicador coloreado.
11(b).2 Material y aparatos.
11(b).2.1 Equipo de valoración potenciométrica equipado con electrodos de vidrio/calomelano o wolframio/calomelano.
11(b).2.2 Agitador magnético.
11(b).2.3 Bureta de 25 ml, dividida en décimas de ml.
11(b).2.4 Pipeta aforada de 25 ml.
11(b).2.5 Matraz aforado de 1.000 ml.
11(b).2.6 Probeta graduada de 50 ml.
11(b).2.7 Matraces redondos de 100 ml provistos de tubo de reflujo, de un metro de longitud, con ajuste normalizado 14/23.
11(b).2.8 Vasos de precipitado de 150 ml, forma alta.
11(b).3 Reactivos.
11(b).3.1 Isopropanol.
11(b).3.2 Etilenglicol. Índice de refracción a 20 °C: 1,42741,4292.
11(b).3.3 Ácido clorhídrico d = 1,18.
11(b).3.4 Hidróxido potásico.
11(b).3.5 Tris- (hidroxi- metil) -aminometano (CH2OH) 3ONH4 de calidad utilizada para la preparación de soluciones patrón, peso equivalente 121,14.
11(b).3.6 Disolución 0,5 N de hidróxido potásico en isopropanol. Pesar 35 g de hidróxido potásico, en lentejas, e introducir en un matraz aforado de 1 litro. Adicionar alcohol isopropílico, agitar con agitador magnético hasta conseguir la disolución completa y enrasar.
11(b).3.7 Disolución 0,5 N de ácido clorhídrico en isopropanol. Medir con probeta 42 ml de ácido clorhídrico y verterlos en un matraz aforado de 1.000 ml, completando el volumen hasta el enrase con isopropanol. Para valorar esta disolución pesar, en vidrio de reloj y con precisión de 0,2 mg, aproximadamente, 1,2 g de tris-(hidroxi-metil)-aminometano, pasándolo a un vaso de 150 ml, forma alta. Disolver en 20 ml de isopropanol, adicionándose 20 ml de etanodiol, efectuándose seguidamente la valoración con la disolución clorhídrica, según se describe en 11 (b).4.2. La valoración también se puede realizar utilizando indicador coloreado, adicionándose para ello 2 gotas de disolución azul de timol al 1 por 100 en isopropanol, acusándose el punto de equivalencia por un viraje brusco del amarillo al rosa. Sean P los gramos de THMAM pesados y V los mililitros de ácido clorhídrico consumidos en la valoración:

11(b).4 Procedimiento.
11(b).4.1 Preparación de la muestra.
La muestra deberá estar seca y libre de materias extrañas en suspensión. En caso contrario, antes de proceder a la pesada, deberá decantarse el agua, si hubiere lugar a ello, y, en todo caso, filtrar por papel de filtro, efectuándose esta operación a una temperatura ligeramente superior a la del punto de fusión de la grasa.
Las grasas que contengan ácidos grasos volátiles no podrán calentarse, debido al riesgo de volatilización de ácidos libres. En estos casos, se procederá a la determinación de la acidez directamente, refiriéndose a muestra seca y exenta de impurezas insolubles, basándose en las determinaciones realizadas sobre muestras independientes.
11(b).4.2 Determinación.
Pesar en un matraz redondo de 100 ml, aproximadamente, 2 g de la muestra. Adicionar seguidamente con pipeta 25 ml de la disolución 0,5 N de 11(b)3.6. Ajustar el tubo de reflujo y calentar hasta ebullición, manteniéndola durante media hora o más si fuera necesario hasta conseguir la saponificación completa. Retirar el matraz y dejar enfriar. Antes de que se enfríe completamente trasvasar el contenido a un vaso de precipitado de 150 ml, lavando el matraz con 30 ml de etilenglicol adicionado en porciones sucesivas. Completar el lavado con 5 ml de isopropanol. Paralelamente se realiza una prueba en blanco con as ml de la disolución de hidróxido potásico 0,5 N. Al terminar el período de ebullición enfriar y transvasar a un vaso de 150 ml como anteriormente. A continuación introducir los electrodos y proceder a la valoración con la disolución de ácido clorhídrico. Ver 11 (b).6.
11(b).5 Cálculos.
Calcular el índice de saponificación aplicando la siguiente fórmula:

Siendo:
Vo = volumen, en ml, consumidos en la valoración en blanco.
V = volumen, en ml, consumidos en el ensayo con la muestra de materia grasa.
N = normalidad de la disolución de ácido clorhídrico.
P = peso, en g, de la muestra.
11(b).6 Observaciones.
Cuando se utilicen electrodos simples la unión entre la disolución saturada de cloruro potásico y la disolución de medida se hará a través de una espiga de porcelana porosa de unos 3 cm de longitud o por cualquier otro sistema que impida una difusión apreciable entre ambas disoluciones durante el tiempo que dure la valoración.
11(b).7 Referencias.
1. Instituto de Racionalización y Normalización del Trabajo. Una Norma Española 55 064.
41. DETERMINACIÓN DE ÁCIDOS GRASOS POR CROMATOGRAFÍA GASEOSA
41.1 Principio.
El método está basado en la separación y determinación por cromatografía gaseosa de los ésteres metílicos de los ácidos grasos.
Es aplicable a aceites y grasas tanto vegetales como animales, que circulan normalmente en el comercio, conteniendo ácidos grasos de 12 a 24 átomos de carbono. En el caso de la mantequilla y de otras grasas que contengan ácidos grasos inferiores, deberá utilizarse un método adecuado para la preparación de los ésteres metílicos o aislamiento de los ácidos libres de pequeña longitud de cadena, siendo necesario para la separación y determinación de estos últimos, efectuar la cromatografía en condiciones distintas de las que se describen en esta norma.
Las condiciones que se especifican no son adecuadas para la determinación de ácidos grasos oxidados y epoxiácidos, por lo que la presencia de estos ácidos dificulta la operación, pudiendo llegar a falsear completamente los resultados.
41.2 Preparación de los ésteres metílicos.
41.2.1 Material necesario.
41.2.1.1 Matraz redondo, fondo plano, de unos 50 ml de capacidad, con boca esmerilada.
41.2.1.2 Refrigerante de agua adaptable al matraz anterior, para su utilización como refrigerante de reflujo.
41.2.1.3 Ampolla de decantación de unos 500 ml de capacidad.
41.2.1.4 Matraz redondo, fondo plano, de unos 100 ml de capacidad.
41.2.1.5 Matraz, fondo plano, de unos 50 ml de capacidad, boca esmerilada, cuello de 40 a 50 mm de longitud y 10 milímetros diámetro exterior.
41.2.2 Reactivos necesarios.
41.2.2.1 Metanol absoluto (99,8 por 100), calidad reactivo para análisis.
41.2.2.2 Sodio metálico reactivo para análisis.
41.2.2.3 Disolución de metilato sódico. Se disuelven 5 g de sodio metal en 1.000 ml de metanol absoluto (0,2 N aproximadamente).
41.2.2.4 Éter de petróleo (p. e. 40° 60 °C) o hexano de calidad adecuada para cromatografía.
41.2.2.5 Disolución en metanol absoluto, de ácido clorhídrico anhidro al 3-4 por 100. Se puede obtener fácilmente el ácido clorhídrico gaseoso haciendo caer lentamente, en aparato adecuado, ácido sulfúrico (d = 1,84), sobre una disolución de ácido clorhídrico (d = 1,16); se seca el gas haciéndolo pasar por un frasco lavador con ácido sulfúrico y se hace llegar al metanol anhidro, contenido en un Erlenmeyer.
Pesando el Erlenmeyer con el metano al comienzo de la operación, por pesadas sucesivas, se determina la cantidad disuelta de clorhídrico, prolongando la operación hasta alcanzar una concentración superior a la deseada. Se adiciona la cantidad de metanol para lograr la concentración del 3-4 por 100 (p/p).
41.2.2.6 Cloruro sódico.
41.2.2.7 Sulfato sódico anhidro, calidad reactivo para análisis.
41.2.2.8 Disolución de fenolftaleína al 1 por 100 en metanol.
41.2.2.9 Disolución de rojo de metilo al 0,1 por 100 en metanol al 60 por 100 (v/v).
41.2.2.10 Gas nitrógeno puro, con un contenido mínimo del 99,8 por 100.
41.2.3 Procedimiento operatorio.
41.2.3.1 Preparación de los ésteres metílicos.‒Los ésteres metílicos de los ácidos grasos pueden ser preparados por interesterificación directa de la grasa, siguiendo el método que se detalla en el párrafo siguiente, el cual tiene un carácter general, aplicable a una grasa, cualquiera que sea su acidez libre y siempre que no tenga un contenido de materia insaponificable superior al 2 por 100, como es el caso de la inmensa mayoría de las materias grasas corrientes.
Pesar 0,3 g de grasa perfectamente homogeneizada en el matraz de 50 ml (ver nota 41.5.1), y se agregan 6 ml de la disolución de metilato sódico. Se coloca el refrigerante al matraz, se hierve hasta obtención de una sola fase y, como mínimo, 5 min. Se interrumpe la calefacción.
Se agregan al matraz 6 ml de la disolución de clorhídrico en metanol y se vuelve a calentar, manteniendo en ebullición durante 5 min. Se enfría y se procede según se indica en el apartado 41.2.3.2 (ver 41.5.3).
41.2.3.1.1 En el caso de materias grasas con una acidez libre no superior al 0,3 por 100, como sucede normalmente en aceites refinados, se puede simplificar el procedimiento omitiendo la metanólisis en medio ácido. Se realiza la metanólisis según se indica en el apartado 41.2.3.1, y, estando todavía caliente el matraz, se agrega, por la parte superior del refrigerante, una gota de disolución indicadora de fenolftaleína y, seguidamente, disolución de clorhídrico en metanol, en una cuantía algo superior a la necesaria para neutralizar el metilato, acusado por el viraje de la fenolftaleína. Se deja enfriar, pasándose la disolución contenida en el matraz a la ampolla de extracción, siguiéndose según se indica en el apartado 41.2.3.2.
41.2.3.1.2 Si se trata de ácidos grasos libres o materias grasas de fuerte acidez (80 por 100 en ácido oleico), se podrá omitir la metanólisis alcalina, reduciendo la operación a la metanólisis en medio ácido. Para ello, se pesa, en el matraz en que se vaya a realizar la operación 0,3 g de muestra, previamente filtrada y seca. Se agregan 6 ml de la disolución de clorhídrico en metano, procediendo como se señala en el apartado 41.2.3.2.
41.2.3.1.3 En el caso de que sea necesario la eliminación previa de la materia insaponificable, se procede según se indica en el método 22(a) «Insaponificable. Método éter de petróleo». La disolución hidroalcohólica de jabón se concentra al vacío, preferiblemente en evaporador rotatorio o bajo corriente de hidrógeno, si no se dispusiese de este aparato, hasta alcanzar unos 50 ml aproximadamente; se pasa el concentrado a una ampolla de extracción, acidificando con ácido clorhídrico 2 N hasta reacción ácida al rojo de metilo. Se extrae con éter de petróleo o hexano, procediéndose de forma análoga a como se indica en el apartado 41.2.3.1.2 para la preparación de los ésteres metílicos.
41.2.3.2 Extracción de los ésteres metílicos.‒Para la extracción de los ésteres metílicos se procede según uno de los métodos que se describen a continuación. En los análisis que interese mucho la rapidez y se opere con aceites y grasas de tipo normal, es preferible el método a). Este método tiene, además, la ventaja de no exigir la evaporación del disolvente y, por tanto, se disminuye el riesgo de pérdida de ácidos grasos de bajo peso molecular, cuando existen en el problema. El método b) es un procedimiento más seguro de recuperación cuantitativa de los ácidos grasos y debe ser utilizado como método de referencia en los límites de aplicación de esta norma. Operando de la forma que se indica en el apartado 41.2.3.2.2, hay riesgo de pérdida de ácidos grasos con longitud de cadena inferior a C12.
41.2.3.2.1 Método a).‒Se pasa la disolución contenida en el matraz a otro de unos 50 ml de capacidad, con cuello estrecho y largo (41.2.1.5), enjuagando con unos 6-8 ml de hexano o heptano de pureza adecuada para cromatografía gaseosa, que se vierten también al matraz, calentándose suavemente sin llegar a hervir, mientras se agita dando un movimiento de rotación al matraz, durante uno o dos minutos. A continuación se agrega disolución acuosa saturada de cloruro sódico en cantidad suficiente para situar la capa de hexano o heptano en el cuello del matraz. Esta disolución, que contiene los ésteres metílicos, debe estar limpia y transparente, tomándose con una pipeta 2 o 3 ml, que se pasan a un frasquito o a una ampolla para su conservación, pudiéndose inyectar directamente esta disolución en el cromatógrafo.
Si la metilación se hubiera efectuado en este matraz, no hará falta el trasvase, adicionándose directamente 6-8 ml de hexano o heptano y la disolución de cloruro sódico.
41.2.3.2.2 Método b).‒Se pasa la disolución contenida en el matraz a una ampolla de extracción de 500 ml, enjuagando con unos 20 ml de éter de petróleo o hexano y añadiendo, a continuación, en la ampolla 100 ml de agua destilada. Se agita enérgicamente y se deja reposar. Se decanta la capa inferior, que se pasa a otra ampolla de extracción y se vuelve a extraer con otros 20 ml de éter o hexano, repitiéndose la operación una vez más. Los tres extractos se reúnen en una ampolla de extracción, y se lavan con porciones sucesivas de 10 ml de agua destilada, hasta eliminación completa del ácido, acusado con la disolución indicadora de rojo de metilo. Se seca la disolución con sulfato sódico anhidro y se elimina el disolvente en el matraz de 100 ml, calentando en un baño de agua bajo corriente de nitrógeno. Esta operación se debe realizar, preferiblemente, en un evaporador rotatorio de vacío.
41.2.3.3 Conservación de la disolución de ésteres metílicos. Los ésteres metílicos obtenidos por uno u otro procedimiento deben ser utilizados en el análisis tan pronto como sea posible. Pueden conservarse durante 24 horas en un frasco bien tapado; desalojando previamente el aire con nitrógeno y guardando a baja temperatura. Para almacenamiento durante períodos de tiempo más largos es necesario conservar en ampolla cerrada a la lámpara, de la cual se ha desalojado previamente, el aire con una corriente de nitrógeno. La eliminación del aire se hace barbotando el nitrógeno en la disolución de hexano.
41.3 Procedimiento cromatográfico.
41.3.1 Material necesario.
41.3.1.1 Cromatógrafo.‒Un cromatógrafo apto para la separación de los ésteres metílicos de ácidos grasos, que disponga de un horno capaz de ser calentado a temperatura regulada hasta 250-300 °C, superior a la temperatura del horno; un sistema de detección sensible, y un aparato registrador continuo.
41.3.1.2 Tubo de nitrógeno.‒Un tubo de nitrógeno a presión, utilizable como gas portador, debiendo tener una riqueza mínima del 99,8 por 100.
41.3.1.3 Jeringa.‒Una jeringa para la inyección de la muestra, con una capacidad de 5 o 10 μl.
41.3.1.4 Aire seco y puro.
41.3.1.5 Hidrógeno con una riqueza mínima del 99,9 por 100 y seco.
41.3.1.6 Columna.
41.3.1.6.1 Columna de acero inoxidable, aluminio, o vidrio de 2-4 mm de diámetro interior y 2 m de longitud rellena de Chromosorb G o W (80-100 mallas), Calite 545 (80-100 mallas) o cualquier otra columna que cumpla las condiciones especificadas en el apartado 41.4.2.
41.3.1.6.2 Preparación de la columna.
41.3.1.6.2.1 Preparación del soporte.‒Para los fines propuestos en esta norma, son recomendables como soportes el Chromosorb G o W de 80-100 mallas, aunque sin excluir la posibilidad de que puedan ser utilizados otros productos análogos de eficacia comprobada, que puedan comportarse de forma análoga o, incluso, superior a los recomendados.
El soporte elegido deberá haber sido lavado con ácido y sometido a un tratamiento de silanización.
Para conseguir un comportamiento óptimo de la columna es importante que el relleno tenga la mayor homogeneidad posible, siendo necesario para ello que el graneado del soporte sea uniforme. En caso de duda, debe procederse a un tamizado del producto lavado con ácido y silanizado, suministrado por la casa fabricante, recogiendo únicamente la porción que pasa por el tamiz 80 y es retenido por el 100 desechando los gruesos y finos (ver 41.5.3).
41.3.1.6.2.2 Impregnación del soporte.‒Se pasa la cantidad de soporte que se desee, introduciéndolo en un matraz de 100 a 200 ml de capacidad, agregando la cantidad necesaria de cloruro de metileno para conseguir una papilla fluida. Se hace el vacío en el matraz hasta que cese el desprendimiento del aire ocluido en el soporte. A continuación se agrega la cantidad de fase fija correspondiente al 2,5 por 100 del peso del soporte utilizado, disuelta en el volumen necesario de cloruro de metileno. Se homogeniza la mezcla, haciendo nuevamente el vacío y manteniéndolo durante 10-15 minutos. Transcurrido este tiempo, se tapa el matraz, dejándolo en reposo durante unas 10-12 horas.
Seguidamente se elimina el disolvente en evaporador rotatorio, con vacío. Una vez seco el relleno, se introduce el matraz en una estufa calentada a unos 150 °C, manteniéndolo así durante 4-6 horas.
Terminada esta operación, el relleno queda listo para su introducción en la columna y acondicionamiento, tal como se describe en los apartados siguientes:
41.3.1.6.2.3 Llenado de la columna.‒Introducir el soporte impregnado con fase fija en la columna, haciendo vacío por un extremo y vibrándola suavemente por percusión o con un vibrador magnético, hasta su llenado total. Es preciso que el material rellene la columna homogéneamente, evitando un empaquetamiento heterogéneo que le haría perder eficacia. Obturar los extremos con tapón de lana de vidrio y/o cilindros de mallas metálicas de cobre o acero inoxidable.
41.3.1.6.2.4 Antes de emplear una columna nueva en la resolución de problemas analíticos, debe ser acondicionada, montándola en el cromatógrafo desconectada del detector y calentando a unos 10° por encima de la temperatura máxima a que vaya a ser utilizada; haciendo pasar, al mismo tiempo, una corriente de nitrógeno, que se mantiene durante veinticuatro horas como mínimo. La columna es apta para su utilización, si la línea base dibujada por el registrador acusa la estabilidad del sistema.
41.3.2 Condiciones operatorias.
41.3.2.1 Se ajusta el flujo del gas portador (nitrógeno), que debe ser el adecuado para permitir la elución del linolenato de metilo en un tiempo mínimo de veinticinco minutos. La presión de entrada y el flujo necesario para conseguirlo varía según la columna y el instrumento utilizado, pero es relativamente constante para un aparato y columna determinada. Es necesario mantener un flujo constante durante todo el análisis. El flujo gaseoso se mide con un medidor de burbuja de jabón y otro dispositivo adecuado.
Se pone en marcha la calefacción del horno de la cámara de inyección y del detector. El horno se regula a una temperatura aproximada de 160°-170 °C; la cámara de inyección, a una temperatura de 50 °C superior a la temperatura de la columna, y el detector, a 25 °C por encima de la temperatura de la columna.
Las condiciones de trabajo indicadas anteriormente deben considerarse como orientación, ya que la imposibilidad práctica de conseguir el mismo comportamiento en columnas diferentes y las variaciones que presentan en sus características y condiciones operativas los diversos aparatos que se encuentran en el comercio, hace imposible fijar, a priori, unas condiciones invariables de trabajo, aplicables a todos los casos y en todas las circunstancias.
Las condiciones operativas más adecuadas deben ser establecidas por cada operador, a la vista de los resultados obtenidos con mezclas patrones de ésteres metílicos, siguiendo las instrucciones que se dan en el apartado 41.3.3.
Para todos los demás detalles operativos no mencionados en esta norma, se deben seguir las instrucciones dadas por la casa fabricante del aparato.
Se prepara una disolución de los ésteres metílicos con acetona o hexano, cuya pureza haya sido previamente comprobada y, utilizando la jeringa, se inyectan 0,4-0,6 μl, y se retira rápidamente la aguja.
El registro obtenido debe satisfacer las condiciones que se indican a continuación; caso contrario, se repite la inyección hasta obtener un cromatograma satisfactorio.
Los requisitos exigibles son los siguientes:
a) El área total descrita en el cromatograma, referida a la sensibilidad máxima utilizada en el curso de la operación, debe ser de un orden aproximado de 2.000 mm2, con una velocidad del papel, en el registrador, de 5mm/min. De esta forma, los componentes presentes, en una cuantía del 0,1 por 100, deben dar un pico como mínimo, de 2 mm2, siendo, por tanto, perfectamente reconocibles
b) Con el fin de conseguir que todos los picos caigan dentro del papel registrador, se utilizará, en cada caso, la atenuación de sensibilidad que sea necesaria.
Una vez conseguido un registro satisfactorio, y habiendo alcanzado nuevamente la pluma la línea base, se interrumpe el funcionamiento del registrador y se retira el papel con el registro para la identificación de los picos cuantitativos.
41.3.3 Identificación de los picos.
41.3.3.1 Criterio basado en los tiempos de retención.‒Refiriéndonos exclusivamente a los ácidos que entran normalmente en la composición de las grasas naturales, sus ésteres aparecen en el cromatograma en orden creciente de sus átomos de carbono y a su insaturación. Esto es, el palmítico H(C16) aparece delante del esteárico (C18), y los ésteres en C18 aparecen en el orden estearato, oleato, linolenato. El éster del ácido aráquico (C20:0), usualmente, aparece antes del linolénico (C18:3) pero, puede ocurrir lo contrario, en algunos casos, dependiendo del tipo de columna y de las condiciones de su utilización; o incluso, superponerse el uno al otro.
Operando en condiciones constantes, los tiempos de retención son reproducibles en cada especie química, siendo el criterio más frecuente empleado para su identificación.
El tiempo de retención viene dado por la distancia, medida en el cromatograma, entre el máximo del pico del aire y la posición del máximo de la banda. Trabajando con detector de llama de hidrógeno, la salida del aire no se detecta, pudiéndose tomar, en este caso, el momento en que se inicia la salida del disolvente, acusada por una fuerte desviación de la pluma del registrador.
41.3.3.2 Criterio basado en los tiempos de retención relativos.‒Los tiempos de retención relativos son más reproducibles. Las retenciones relativas vienen determinadas por el cociente de dividir el tiempo de retención de cada pico por el tiempo registrado para el pico del palmitato de metilo, o bien por otro éster que se tome como comprobación, determinados todos ellos según el criterio expuesto en el apartado 41.3.3.1.
41.3.3.3 Como el comportamiento de la columna cambia como consecuencia de factores muy diversos, y durante su utilización continuada experimenta un proceso de envejecimiento que altera su capacidad de retención, es conveniente comprobar, periódicamente, la posición de los distintos ésteres en el cromatograma, utilizando mezclas patrones convenientemente preparadas, o bien ésteres metílicos de una grasa previamente analizada y conocida. Esto constituye una de las operaciones de comprobación a que se hace referencia en el capítulo 41.4.
41.3.4 Determinación cuantitativa.
La determinación cuantitativa se basa en el principio de que los pesos de cada uno de los componentes separados en la mezcla son proporcionales a las áreas comprendidas dentro de los triángulos dibujados debajo de cada pico. El área de cada triángulo se obtiene trazando rectas tangentes a los puntos de inflexión de cada pico, prolongándolas hasta su intersección con la línea base y multiplicando la altura del triángulo por la mitad de la base. En el caso de haber trabajado con atenuaciones diferentes para cada pico, se referirán todas las medidas a una misma sensibilidad del registrador, multiplicando la altura por el factor de atenuación correspondiente en cada caso, y el valor de la altura así corregida, por la mitad de la base. Si no es necesario efectuar corrección en relación al cambio de atenuación, como ocurre en la mayor parte de los análisis de rutina en que se trabaja con una sola atenuación, la medida del área se realiza más cómodamente multiplicando la altura del pico por el ancho a la mitad de la altura. El contenido de cada ácido en la muestra viene dado por la expresión

siendo A el éster metílico correspondiente a un pico dado.
41.4 Operaciones de comprobación.
41.4.1 Reactivos necesarios:
Laurato de metilo.
Palmitato de metilo.
Estearato de metilo.
Oleato de metilo.
Linoleato de metilo.
Estos ésteres metílicos han de ser puros, comprobada su pureza por cromatografía gaseosa, y conservados en condiciones que garanticen su inalterabilidad y comerciales como se indica en 41.2.3.3.
41.4.2 Prueba de comportamiento del instrumento y de la columna.
Se realiza determinando la resolución de dos productos críticos, como son el oleato y el estearato de metilo. La resolución viene determinada por la expresión:

Siendo:
D = distancia entre los dos máximos de los picos del oleato y el estearato.
O = ancho de la base del pico correspondiente al oleato.
E = ancho de la base del pico correspondiente al estearato.
Estos valores se determinan sobre el cromatograma obtenido con una muestra que contenga cantidades aproximadamente iguales de estearato y oleato de metilo, inyectando una cantidad tal que la altura de estos picos alcance al 25-50 por 100 del ancho del papel de registro.
Si la resolución calculada es igual o mayor que 1,0, la columna y el instrumento se encuentran en condiciones satisfactorias. Todas las columnas en el transcurso de su utilización sufren una pérdida gradual en la resolución de los picos; cuando el valor llegue a ser inferior a 1,0, debe instalarse una nueva columna.
41.4.3 Situación de los ésteres en el cromatograma.
Utilizando los productos patrones a que se hace referencia en el apartado 41.4.1, se prepara una mezcla que contenga, preferiblemente, cantidades de un orden aproximado al existente en la grasa o grasas que se trata de analizar. Se registra el cromatograma de la forma usual, determinando los tiempos de retención absolutos o relativos para cada una de las especies contenidas en la mezcla.
En el caso de tener que identificar en el problema algún pico que no coincide con los ésteres contenidos en la mezcla patrón, es de suma utilidad efectuar, a partir de los datos obtenidos con la mezcla patrón, la representación gráfica de la función que relaciona los logaritmos de los tiempos de retención, con el número de átomos de carbono de cada ácido; para los términos comprendidos en una misma serie homóloga, esta función es lineal; por lo tanto, los tiempos de retención de los términos de la serie de los que no se disponga de muestra patrón pueden calcularse por interpolación o viceversa.
41.4.4 Calibrado para aplicación cuantitativa.
Utilizando los productos patrones a que se hace referencia en el apartado 41.4.1, se prepara una mezcla que contenga cantidades exactamente pesadas de cada uno de los ésteres, debiendo tener una composición análoga a la de la muestra problema. Se registra el cromatograma de la forma usual, efectuándose los cálculos cuantitativos según se indica en el apartado 41.3.4.
Los resultados deducidos del cromatograma coinciden, normalmente, con los valores reales de la mezcla patrón. Sin embargo, en algunos casos, debido a diversas causas, se observan discrepancias que pueden ser corregidas aplicando factores de corrección. Estos factores de corrección no son aplicables más que en la parte lineal de la curva de respuesta del detector para cada constituyente. Por consiguiente, el operador debe ser extremadamente prudente en lo que respecta a la aplicación de estos factores, ya que ellos pueden variar en función de la composición de la mezcla a analizar, modificaciones en el detector y en el amplificador, alteración de la fase fija, etc. Si la cantidad encontrada para un ácido graso cualquiera de la mezcla patrón discrepa en más de un 10 por 100 de la cantidad calculada, éste indica que el aparato no funciona correctamente, siendo necesario buscar la causa.
El factor de corrección para cada ácido se calcula con relación al ácido palmítico, utilizando mezclas patrones con una composición análoga a la de la muestra problema que se trata de analizar. Se procede de la forma siguiente: se divide el porcentaje de cada ácido por el área del pico correspondiente y el cociente por el valor obtenido para el ácido palmítico:

Siendo:
fx = factor de corrección del ácido.
x = tanto por ciento del ácido en la mezcla patrón.
Ax = área del pico x.
P = tanto por ciento del ácido palmítico en la mezcla patrón.
Ap = área del pico del ácido palmítico.
Para aplicar estos factores a la mezcla problema se multiplican por la relación entre el área medida para cada ácido en el cromatograma y el área del pico correspondiente al ácido palmítico; se obtiene, procediendo de esta forma, la relación entre el contenido de cada ácido y el palmítico, tomado como unidad.

Si están comprendidos en el cromatograma todos los ácidos componentes de la mezcla problema, a partir de las relaciones calculadas se puede pasar fácilmente a la composición centesimal.
41.5 Notas.
41.5.1 En el caso de utilizarse el método de extracción a) (apartado 41 2.3.2.1), la pesada y metilación de la muestra se efectuará más cómodamente en el matraz de cuello largo, descrito en el apartado 41.2.1.5, efectuándose en el mismo recipiente, sin necesidad de trasvase, la adición de la disolución saturada de cloruro sódico.
41.5.2 En el caso de que se tuviese dificultad en la preparación de la disolución de ácido clorhídrico, se podría sustituir con una disolución del 5-6 Por 100 (p/p) de ácido sulfúrico (d = 1,04) en metanol anhidro utilizable también en las operaciones descritas en los apartados 91.2.3.1.1 y 41.2.3 1.2.
41.5.3 La designación de los tamices corresponde a la nomenclatura del sistema U. S., referida a número de mallas por pulgada lineal: de acuerdo con la Norma UNE 7050, los tamices de 80 y 100 mallas/pulgada lineal (sistema U. S.) serían designados respectivamente, tamiz 0,177 UNE 7050 y tamiz 0,149 UNE 7050, siendo las cifras indicadas las aberturas de malla expresadas en milímetros.
41.6 Referencias.
UNE 55.037 Materias grasas. Determinación de ácidos grasos por cromatografía gaseosa.
43. RECONOCIMIENTO DE ÉSTERES NO GLICÉRIDOS EN GRASAS COMESTIBLES POR CROMATOGRAFÍA EN CAPA FINA
43.1 Principio.
Disolución de la muestra en hexano y posterior separación de los triglicéridos por cromatografía en capa fina.
Es aplicable a todos los aceites vírgenes o refinados, utilizables directamente en la alimentación humana.
El método ha sido estudiado con ésteres de ácidos grasos y los alcoholes siguientes: metanol, 1,2 y 1,3 propilenglicol y etilenglicol.
Los límites detectados, según ensayo colaborativo en el que han intervenido cinco laboratorios, oscila entre 0,7 por 100 (p/p) y 1 por 100 (p/p).
43.2 Material y aparatos.
43.2.1 Equipo de cromatografía en capa fina, compuesto de placas de gel de sílice G y un espesor de capa de 0,25 mm o de 0,40 mm en caso de ser necesaria una mayor resolución.
43.2.2 Placa calefactora adecuada para quemar placas que permita alcanzar temperaturas de 360 °C.
43.2.3 Microjeringa de 10 μl.
43.3 Reactivos.
43.3.1 Hexano para cromatografía (d20 ºC = 0,684).
43.3.2 Éter etílico (d20 ºC = 0,715).
43.3.3 Líquido de desarrollo.
Mezclar 92 volúmenes de hexano y 8 volúmenes de éter etílico.
43.3.4 Ácido sulfúrico al 50 por 100.
43.4. Procedimiento.
Depositar con una jeringa 2 a 3 μl de la disolución de la muestra en hexano al 10 por 100, aproximadamente, sobre la placa de cromatografía, a una distancia aproximada de 1 cm del borde inferior de la capa de sílice.
Introducir en la cubeta contenida el líquido de desarrollo hexano-éter etílico, esperando hasta que el frente del disolvente se sitúe a unos 3 cm, aproximadamente, del borde superior de la placa. Sacar la placa de la cubeta y dejar secar al aire, lo cual se consigue en unos minutos. Con el fin de facilitar el reconocimiento de los ésteres más difícilmente separables, es recomendable y necesario en algunos casos efectuar dos o tres desarrollos. Para ello, terminado el primer desarrollo y una vez seca la placa, volver a introducir en la cubeta manteniéndola hasta que el frente del disolvente se haya situado a la misma altura anterior. Repetir el proceso una vez más si es necesario. En los casos de duda sobre el resultado positivo de la prueba, deberá repetirse la cromatografía, sometiendo la placa a dos o tres desarrollos.
Pulverizar la placa seca con una disolución de ácido sulfúrico al 50 por 100 y quemar sobre la placa calefactora a unos 300 °C.
43.5 Interpretación de resultados.
A un tercio, aproximadamente, del borde inferior de la placa aparece una mancha intensa correspondiente a los triglicéridos. En aceites puros no aparece por encima ninguna otra mancha próxima a la de los triglicéridos, a excepción de algunos componentes del insaponificable que marchan con el frente del disolvente. Una mancha, más o menos intensa, situada por encima, próxima a la de los triglicéridos, acusa la presencia de ésteres extraños al glicerol. La distancia relativa entre estas dos manchas depende, lógicamente, del alcohol de que se trate; los ésteres de monoalcoholes, tales como el metílico o etílico, se sitúan más distanciados de como lo hacen los ésteres de dialcoholes, tales como el etilenglicol o propilenglicol.
43.6 Referencias.
1. Instituto de Racionalización del Trabajo. Una Norma Española 55.085.
44. TEMPERATURA DE INFLAMACIÓN
44.1. Principio.
Inflamación momentánea de los vapores desprendidos de la materia grasa en ensayo, en contacto con el aire, operando en condiciones determinadas.
44.2 Material y aparatos.
44.2.1 Aparato de Pensky-Martens en taza cerrada (fig. 44.1) que consta de los elementos siguientes:
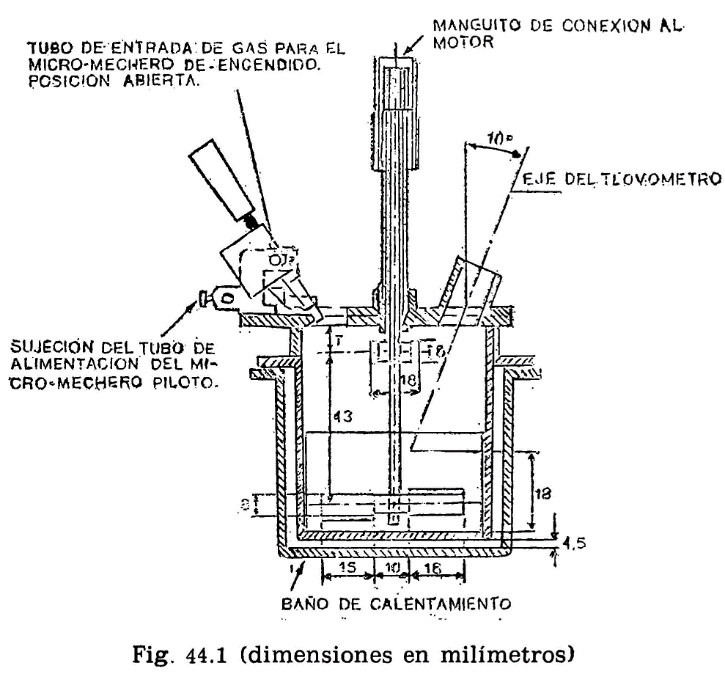
44.2.1.1 Taza cilíndrica de latón o bronce o aleación similar no oxidable y de conductividad térmica equivalente (figura 44.2). Deberá ir provista de una pestaña a todo su alrededor para su ejecución en el baño de calefacción, debiendo, además, fijarse en una posición determinada, impidiendo todo movimiento giratorio una vez situada dentro del baño. Llevará una marca circular alrededor de la pared interior, a una altura de 34 milímetros del fondo, indicando la cantidad que debe tomarse del aceite a ensayar. El volumen necesario para el ensayo, señalado por esta marca es de 70 ml. Llevará también un mango con el aislamiento conveniente, que permita el manejo cómodo de la taza para su colocación y retirada del bloque.
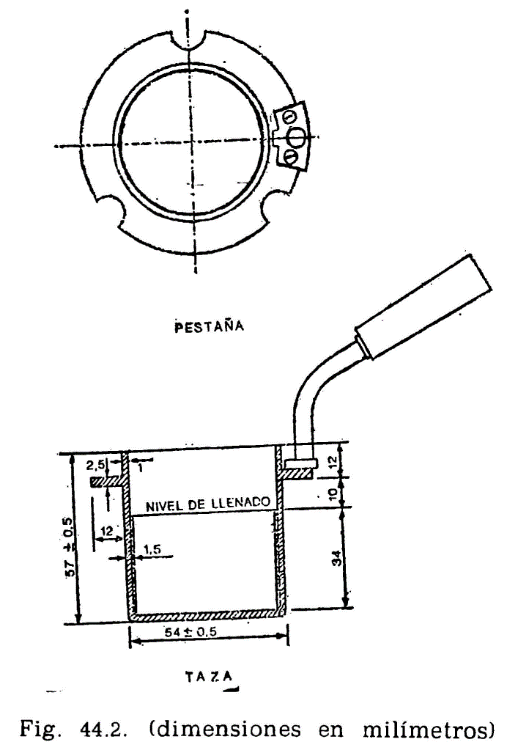
44.2.1.2 Tapa (fig. 44.3).
La taza irá provista de una tapa, la cual encajará en la parte superior, estando provista de cuatro orificios cuya situación y dimensiones son las indicadas en la figura.
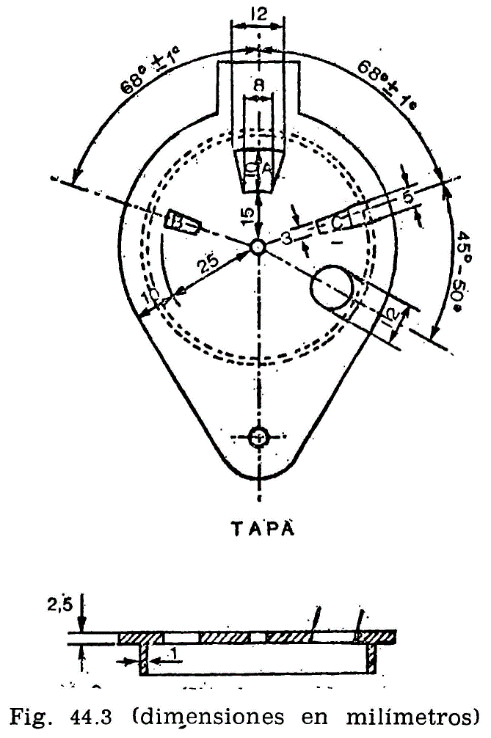
44.2.1.3 Obturador (fig. 44.4).
Encima de la tapa lleva una lámina que puede girar sobre el eje central del aparato, pudiendo efectuarse este giro accionando manualmente un mecanismo que permita el desplazamiento de la lámina entre dos posiciones extremas: Una, que puede denominarse posición de reposo, y otra, posición de encendido. El mecanismo irá provisto de un muelle que mantenga el obturador en posición de reposo, siendo necesario actuar en sentido contrario para pasarlo a la posición de encendido; de la presión manual, el obturador deberá recobrar automáticamente la posición inicial.
En la posición de reposo, las tres aberturas A, B y C de la tapa permanecen cerradas por el obturador; al girar la tapa, accionando el mecanismo aludido, y llevarla a la posición opuesta, se abrirán las tres aberturas, haciendo posible, al mismo tiempo, los movimientos que se indican en los dos apartados siguientes referentes al micromechero de encendido y a la agitación.
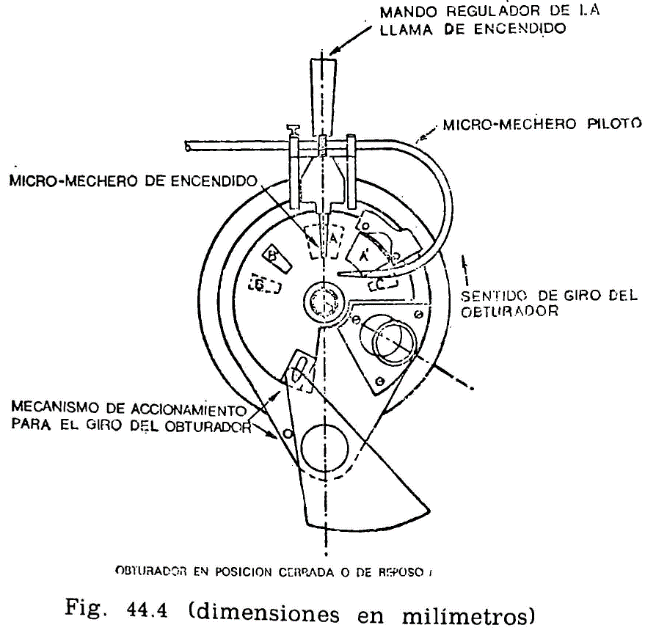
44.2.1.4 Micromechero de encendido (fig. 44.4).
Está constituido por un pequeño tubo, con un orificio de salida de 0,5 milímetros de diámetro, provisto de una válvula accionada por un tornillo que permite la regulación de la llama. Puede utilizarse gas ciudad o butano.
El micromechero va montado en un eje transversal, que permita un movimiento basculante, pudiendo introducirse la punta del mechero por el orificio A de la tapa (fig. 44.3) apoyándose en el borde. Situado en esta posición, quedarán introducidos dos milímetros de mechero y en una posición inclinada formando un ángulo de 40 grados con la vertical.
44.2.1.5 Micromechero piloto.
Es un mechero de características análogas al anterior, pero con un orificio de salida de 0,25 milímetros y sin válvula de regulación. Está situado en una posición horizontal, perpendicularmente al mechero de encendido, separado dos milímetros de la tapa móvil designado como obturador (fig. 44.4). El objeto de este micromechero es el mantener encendida una llama piloto que, al bascular el micromechero de encendido, e inmediatamente antes de penetrar en la abertura A, se encienda, penetrando ya encendido; al volver el mechero a su posición normal horizontal deberá apagarse nuevamente.
44.2.1.6 Agitador.
La tapa descrita anteriormente irá equipada con un agitador (fig. 44.1) montado en el centro de la tapa y constituido por dos láminas rectangulares de 15 mm de longitud y 8 mm de ancho, dispuestas formando entre sí un ángulo de 90°; la distancia entre los extremos de las láminas será de 40 mm y la distancia entre la cara interior de la tapa y el extremo inferior del agitador será de 50 mm. En la parte superior del eje y en la posición que se indica en el dibujo, llevará un segundo sistema de paletas, con un ancho de 8 mm y una distancia entre los extremos de las láminas de 18 mm. El eje con los sistemas de paleta irá conectado a un motor eléctrico, verificándose esta conexión mediante un cable flexible que pueda conectarse y desconectarse a voluntad.
El mando de accionamiento del agitador irá unido a un sistema mecánico adecuado para que al mismo tiempo de accionar el obturador, desconecte al motor del agitador y haga vascular el micromechero de encendido, para encender la llama e introducirla en la taza, tal como se ha explicado anteriormente: al dejar en libertad el mando de accionamiento del obturador, se establecerá la situación anterior, quedando apagado el micromechero en su posición normal, cerrada la taza y restablecido el funcionamiento del motor. El ciclo completo de estos movimientos deberá realizarse en un tiempo comprendido entre dos y tres segundos.
44.2.1.7 Baño de calentamiento.
Está constituido por un cilindro metálico de aleación análoga a la de la taza, provisto de un sistema de calefacción en el que puede utilizarse una llama o calentamiento eléctrico, lo cual es preferible. En cualquier caso, el sistema utilizado dará lugar a un calentamiento uniforme de toda la superficie del baño, tanto en el fondo como en las paredes laterales, y la fuente de calefacción tendrá potencia suficiente para elevar la temperatura del baño de la forma que se especifica más adelante, pudiendo llevar el aceite contenido en la taza del ensayo a una temperatura de 350 °C. El baño irá provisto, en su parte superior, de una placa con un orificio de 54,5 mm de diámetro, por el que se introducirá la taza conteniendo el aceite a ensayar, haciendo descansar sobre la placa la pestaña de que va provista la taza (fig. 44.2). La posición de la taza se fija exactamente en el centro del baño mediante unos tornillos que encajan en unas muescas situadas en la pestaña o utilizando cualquier otro dispositivo de fijación, quedando espacio libre entre la taza y el baño de calentamiento, de 4,5 mm, que debe mantenerse uniforme por toda la superficie (fondo y paredes laterales).
44.2.1.8 Termómetros.
Adaptables al aparato, con intervalos de 1 °C y comprendiendo los límites entre los cuales se presume se han de situar las temperaturas a medir. Deberán haber sido debidamente contrastados, con una tolerancia en el error de escala de ±0,5 grados centígrados.
44.2.2 Centrífuga de cabeza oscilante, con capacidad suficiente para centrifugar, en una sola operación, unos 100 ml de aceite.
44.3 Reactivos.
44.3.1 Sulfato cúprico anhidro, químicamente puro. Pulverizar en un mortero unos 50 g de SO45H2O. El producto pulverizado se coloca en una cápsula que se mantiene en una estufa dos horas a 150 °C.
44.4 Procedimiento.
44.4.1 Preparación de la muestra.
Tomar aproximadamente 100 g de muestra y adicionar un 5 por 100 de su peso de sulfato cúprico anhidro, agitar durante un minuto en una vasija cerrada y dejar reposar durante media hora. Centrifugar en tubo cerrado con tapón esmerilado a 2.800 r.p.m., debiendo quedar el aceite limpio (ver 44.6.1 y 44.6.2).
44.4.2 Ensayo preliminar.
En el caso general de tratarse de una muestra para la que se desconozca totalmente su comportamiento en el ensayo, será necesario efectuar previamente una o varias determinaciones preliminares que sirvan para conocer el valor aproximado del «punto de inflamación» Si se tratase de una muestra de naturaleza conocida para la que pueda preverse el orden aproximado de «la temperatura de inflamación», podría omitirse el ensayo preliminar, pudiendo cumplirse los requisitos exigidos en el ensayo definitivo.
44.4.3 Ensayo definitivo.
Llenar la taza del aparato con la grasa convenientemente preparada, cuidando que la base del menisco coincida con la línea marcada en el interior y evitando la formación de burbujas de aire. Colocar la tapa así preparada en el aparato, junto con los demás dispositivos y el termómetro de escala adecuada.
Encender la llama del micromechero piloto ajustando la entrada de gas de forma que la longitud de la llama sea de unos 4 mm; ajustar también la entrada de gas en el micromechero de encendido para que la llama, al efectuar el disparo, tenga una longitud de 5 a 6 mm.
Poner en marcha el dispositivo de calefacción y el agitador, regulado de forma que no sobrepase una temperatura que se sitúe de 5 a 10° por bajo de «la temperatura de inflamación prevista»; el agitador deberá girar a una velocidad de 60 a 126 revoluciones por minuto.
Alcanzada la temperatura previamente establecida, continuar la calefacción, regulada de forma que el aumento de temperatura sea de 0,5 °C/min. Cuando falten unos 3 °C para alcanzar la «temperatura de inflamación» se comienzan los ensayos. Para ello y a intervalos de un minuto se interrumpe la agitación y se acerca la llama a la superficie del aceite, abriendo el obturador que cierra la ventana de la taza y vasculando el micromechero hasta introducir la llama en el interior, debiéndose realizar esta operación en medio segundo. Dejar en esta posición un segundo, volviéndolo rápidamente a su posición primitiva, poniendo inmediatamente en marcha la agitación (ver 44.6.4).
Repetir esta operación a intervalos de un minuto, hasta que se observe una inflamación clara pero fugaz en los vapores que existen en el interior de la taza, y se extienden por toda la superficie del aceite. La temperatura a la que se produce este fenómeno es la que se toma como «temperatura de inflamación». Anotar la presión atmosférica a la que se ha realizado el ensayo (ver 44.6.5 y 44.6.6).
El número de aperturas del obturador hasta alcanzar la «temperatura de inflamación» no deberá ser nunca superior a cinco. Si fuera necesario rebasar esta cifra, repetir el ensayo tomando una nueva muestra de aceite.
Análogamente, si se consiguiese la inflamación en el primer intento, la temperatura registrada no se dará como aceptable, debiéndose repetir el ensayo con las correcciones oportunas.
La duración total del ensayo será aproximadamente de una hora y media.
44.5 Cálculos.

Siendo:
Te = Valor leído como temperatura de inflamación.
Tc = Valor corregido, expresado en grados centígrados, referido a la presión normal.
p = Presión atmosférica, expresada en mm de Hg, a la que se ha efectuado la medida.
La diferencia entre resultados sucesivos obtenidos por el mismo operador, con el mismo instrumental y con una misma muestra, no debe sobrepasar dos unidades. De no ser así, realizar un tercer ensayo o los que fueran necesarios hasta alcanzar la constancia debida.
44.6 Observaciones.
44.6.1 Si la grasa es sólida a la temperatura del laboratorio, fundir calentando a una temperatura que no exceda de 10 °C a la temperatura de fusión. La determinación de la temperatura de inflamación se comenzará, en este caso, a la temperatura a la cual se haya realizado este proceso previo de fusión. Si fuese necesaria la desecación, se realizará, también, sobre la grasa licuada.
44.6.2 La deshidratación puede ser suprimida si la humedad de la muestra no es superior a 0,1 por 100. Si el contenido en agua fuese más elevado, la desecación es necesaria para eludir la posibilidad de que se forme una espuma excesiva, que podría apagar la llama al introducir el micromechero en la taza.
44.6.3 En los aparatos de funcionamiento automático, al reaccionar el mecanismo del obturador, se producen simultáneamente todos los movimientos descritos, debiéndose preocupar el operador únicamente de mantener el mecanismo en tensión durante el tiempo establecido de un segundo, observando el proceso de inflamación y la temperatura.
44.6.4 El intervalo de un minuto entre cada dos ensayos de inflamación es el tiempo mínimo que se considera necesario para restablecer la concentración de saturación de los vapores del espacio libre de la taza, en equilibrio con el aceite en ensayo, equilibrio éste que se altera al efectuarse cada ensayo de inflamación.
44.6.5 El verdadero fenómeno de inflamación no debe confundirse con el halo azulado que se observa algunas veces rodeando la llama; esto indica que el fenómeno de inflamación está próximo, pero no debe confundirse con la verdadera inflamación del aceite.
Si al introducir la llama de encendido por la abertura de la placa se produce una inflamación permanente de los gases combustibles distinta al destello que se ha descrito anteriormente, este hecho pone en evidencia que el aceite se encuentra a una temperatura superior a su punto de inflamación, debiendo repetirse la experiencia partiendo de una nueva muestra.
44.7 Referencias.
1. Instituto de Racionalización del Trabajo. Una Norma Española. 55.103.
45. RECONOCIMIENTO DE ANTIOXIDANTES
45.1 Principio.
Los antioxidantes son extraídos por el acetonitrilo de una solución de la muestra en hexano y posterior fraccionamiento e identificación.
Aplicable a materias grasas destinadas directamente a la alimentación humana. No aplicable a grasas brutas o a aquellas otras que introduzcan impurezas en el extracto que dificulten el fraccionamiento cromatográfico y el reconocimiento de antioxidantes.
Los antioxidantes identificados por este método son: Ácido nordihidroguayarético (NDHG) galato de propilo (GP), galato de octilo (GO), galato de dodecilo (GD), butilhidroxianisol (BHA) y butilhidroxitolueno (BHT).
45.2 Material y aparatos.
45.2.1 Equipo de cromatografía en capa fina, con placas de gel de sílice G de tamaño conveniente, según el número de muestras que se deseen examinar en una sola operación y un espesor de capa de 0,25 milímetros. Ver 45.6.1.
45.2.2 Evaporador rotatorio.
45.2.3 Matraz de fondo redondo de 250 ml, utilizable con el evaporador rotatorio.
45.2.4 Estufa de desecación con calefacción eléctrica y regulación de temperatura, con un error de ± 2 °C en todo el ámbito interior de la estufa.
45.2.5 Ampollas de extracción con capacidad de 250 ml.
45.2.6 Matraces cónicos de 250 ml y 1.000 ml con tapón esmerilado y cono normalizado 29/32.
45.2.7 Matraces aforados de 100 ml con tapón esmerilado y cono normalizado 14/23.
45.2.8 Vaso de 150 ml.
45.2.9 Jeringa de 20 μl, graduada en μ.
45.3 Reactivos.
45.3.1 Gel de sílice G, de calidad adecuada para cromatografía en capa fina y en especial para el fraccionamiento de los productos que se trata de separar, lo que se comprueba con los patrones correspondientes.
45.3.2 Metanol con un contenido de agua que no sobrepase el 0,5 por 100 (v/v).
45.3.3 Etanol con una riqueza de 96/97 por 100 (v/v). 45.3.4. Hexano normal para cromatografía (d20° = 0,584).
45.3.5 Acetonitrilo.
45.3.6 Acetato de etilo.
45.3.7 Benceno.
45.3.8 Cloroformo.
45.3.9 Ácido acético cristalizable.
45.3.10 Acetonitrilo saturado de hexano.‒En un matraz cónico de 100 ml introducir 900 ml de acetonitrilo. Agregar 100 ml de hexano y un poco de sulfato sódico seco. Agitar, cerrar el matraz con el tapón y dejar en reposo unas doce horas. En el momento del uso, sacar la cantidad necesaria de acetonitrilo
45.3.11 Hexano saturado de acetonitrilo.‒Proceder como se indica anteriormente, invirtiendo las cantidades de los dos disolventes. Conservar en el matraz hasta su utilización.
45.3.12 Líquido de desarrollo.‒Inmediatamente antes de su utilización preparar una mezcla de hexano, benceno y ácido acético en las proporciones 40:40:20 (v/v/v).
45.3.13 Revelador.‒Solución al 1 por 100 en etanol (m/v) de dicloro-2-5-quinona clorimida.
45.3.14 Soluciones patrón.‒Disolver al 0,1 por 100 (m/v) en etanol los antioxígenos cuya identificación se quiera realizar en la grasa. Estos productos deberán ser puros, comportándose en la cromatografía como una sola especie química; si apareciesen impurezas, éstas no deberán perturbar la marcha del proceso ni el reconocimiento de los otros antioxidantes.
45.4 Procedimiento.
45.4.1 Extracción de los antioxidantes.
Pesar de 7,5 a 10 ml de la muestra de aceite o grasa e introducirla en un vaso de 150 ml. Disolver con 100 ml de hexano, pudiéndose calentar suavemente en caso de necesidad para acelerar el proceso de disolución. Pasar la solución a una ampolla de extracción de 250 ml. Lavar el vaso con 25 ml de hexano, agregando este líquido a la ampolla de extracción. No debe quedar ninguna partícula insoluble en la solución. Añadir 25 ml de acetonitrilo saturado de hexano y agitar durante un minuto. Sacar la fase inferior de acetonitrilo, pasándola a una segunda ampolla de 250 ml. Si se forma una emulsión, calentar suavemente la ampolla haciendo caer sobre ella, desde un frasco lavador, un chorro de agua a unos 50 °C, haciéndola girar al mismo tiempo muy lentamente. Prolongar la operación hasta lograr la separación en dos fases limpias.
Repetir otras tres veces consecutivas la extracción con el acetonitrilo saturado de hexano, empleando 25 ml en cada extracción acumulando los extractos en la misma ampolla.
Los extractos reunidos de acetonitrilo se lavan dos veces con hexano saturado de acetonitrilo, empleando 25 ml en cada lavado.
Pasar el extracto en acetonitrilo, una vez lavado, a un matraz de fondo redondo de 250 ml. Evaporar el disolvente en evaporador rotatorio, con vacío, calentar muy cuidadosamente y evitando que la temperatura del baño de agua no sobrepase, en ningún momento, los 40 °C.
Una vez evaporado el disolvente, disolver el residuo en 2 ml de metanol, pasando la disolución a un frasquito de capacidad adecuada para su conservación y utilización en la cromatografía.
Si el residuo obtenido en la concentración no se disuelve totalmente en el metanol, filtrar la solución.
Para contenidos pequeños de antioxidantes, por ejemplo, de un orden inferior a 0,1 por 1.000, debe emplearse menos disolvente.
45.4.2 Cromatografía en capa fina.
Depositar 10 μl de la disolución en metanol de los antioxidantes en una placa de dimensiones adecuadas, previamente activada en las condiciones recomendadas para cada caso, y en el centro de una línea situada a unos 2 cm del extremo inferior de la placa.
Depositar 4 μ, de cada uno de los patrones con que se desee operar, a la derecha y a la izquierda de la mancha problema, distantes entre sí 10-15 mm.
Introducir la placa en la cubeta de desarrollo, cuidando que las manchas depositadas queden por encima del nivel del líquido en la cubeta.
Dejar la cubeta preferiblemente en la oscuridad o en una estancia débilmente iluminada, prolongando el desarrollo hasta que el frente del disolvente se sitúe a unos 15 cm de la línea de partida de los antioxidantes. Sacar la placa dejándola secar al aire.
Pulverizar la placa con el revelador, introduciéndola después en una estufa de aire regulada a 100 °C (+ 2 °C), donde permanecerá diez a quince minutos.
Sacar la placa de la estufa y observar las manchas del problema, comparándolas con las de los patrones.
45.5 Interpretación de resultados.
El orden de colocación de los antioxidantes, en orden de menor a mayor desplazamiento, es el siguiente: NDHG, GP, GO, BHA, BHT (ver 45.6.2 y 45.6.3).
45.6 Observaciones.
45.6.1 En determinados casos, para conseguir una separación efectiva de antioxidantes, con valores de Rf muy próximos, como por ejemplo el NDHG y GP, es aconsejable operar con espesores de gel de sílice de 0,40 mm en lugar de 0,25 mm, que es el más frecuente y con el que se consiguen normalmente resultados satisfactorios.
45.6.2 Se puede intensificar la coloración de las manchas, facilitando su reconocimiento y la sensibilidad del método, mediante reacción con el amoníaco.
Para ello, la placa, una vez revelada y seca, se introduce en una cubeta saturada de vapores de amoníaco, manteniéndola allí hasta observar una intensificación del color de las manchas. Se adquieren coloraciones características en algunos casos.
Debe cuidarse no prolongar demasiado el contacto con el amoníaco, en evitación de que se coloree el fondo de la placa, lo cual, en vez de beneficiar, podría dificultar el reconocimiento de los antioxidantes.
45.6.3 A título de orientación, se dan a continuación los valores Rf obtenidos con los seis antioxidantes citados anteriormente:
| Antioxidantes. | NDHG | GP | GO | GD | BHA | BHT |
| Rf. | 0,10 | 0,14 | 0,28 | 0,35 | 0,75 | 0,98 |
45.6.4 La aparición de una mancha de BHT muy débil, en comparación con el patrón, puede ser debido a una pérdida del producto en el lavado con hexano saturado de acetonitrilo. En este caso, proceder como sigue: Realizar la extracción tal y como dice el método. Recoger por separados los líquidos procedentes de la extracción con acetonitrilo saturado de hexano y los que proceden del lavado con hexano saturado de acetonitrilo. Evaporar los disolventes. Disolver ambos residuos con metanol (2 ml). Sobre la placa depositar unos 10 μl del extracto procedente de la extracción con acetonitrilo y 15 μl del que procede de los lavados con hexano. De esta forma, el BHT se detecta en ambos desarrollos, pudiéndose considerar la cantidad presente en la muestra por comparación con el patrón como «suma» de las intensidades de ambas manchas.
45.7 Referencias.
1. Instituto de Racionalización del Trabajo. Una Norma Española. 53.017.
46. ÁCIDOS GRASOS DE CADENA CORTA
46.1 Principio.
Obtención de los ésteres metílicos de los ácidos grasos mediante reacción con una solución de hidróxido potásico en metanol y subsiguiente inyección directamente de la disolución de ésteres metálicos en el cromatograma.
El método es aplicable a las grasas de mantequillas u otras que contengan ácidos grasos de longitud de cadena inferior al C14 y siempre que el contenido de ácidos libres no exceda del 1 por 100 expresados en ácido oleico.
46.2 Material y aparatos.
46.2.1 Matraces con boca esmerilada y fondo redondo de 50 y 100 ml de capacidad.
46.2.2 Pipetas aforadas de 1 ml, 2 ml y 10 ml.
46.2.3 Matraces aforados de 50 y 100 ml de capacidad.
46.2.4 Probeta graduada de 10 ml.
46.2.5 Jeringa de características adecuadas para la inyección de la muestra, graduada en décimas de ml, con una capacidad total de 1 a 10 ml.
46.2.6 Cromatógrafo apto para trabajar en fase gaseosa, provisto de horno capaz de ser calentado hasta 250º-300 °C y sistema de regulación que permita controlar la temperatura con un error de ±1,0 °C. Equipado con programador de temperatura capaz de llevar la temperatura del horno de 60 °C a una velocidad de 4 °C/min. Provisto de regulación independiente de la temperatura del inyector, que podrá ser calentado a una temperatura superior, por lo menos, en 50° a la máxima alcanzable por el horno provisto de un sistema de detección sensible, de ionización de llama de hidrógeno, que pueda ser mantenido a la temperatura de la columna, a unos 50 °C por encima de la del horno.
46.2.7 Registrador con una tensión de entrada adecuada a la salida del amplificador del cromatógrafo, con una velocidad de respuesta mínima capaz de producir la deflexión completa de la escala en un segundo y una velocidad de desplazamiento del papel de 5 mm/min, que permita la posibilidad de variar esta velocidad acelerando o retardando el desplazamiento.
46.2.8 Tubo de nitrógeno a presión utilizable como gas portador, debiendo tener una riqueza mínima del 99,8 por 100.
46.2.9 Tubos de hidrógeno y aire a presión necesarios para el caso en que se utilice detector de llama de hidrógeno. El hidrógeno deberá tener una riqueza mínima del 99,8 por 100, debiendo estar seco. Como medida de seguridad, es muy conveniente colocar a la entrada de los gases en el cromatógrafo sendos tubos de desecación provistos de criba molecular 13X.
46.2.10 Columna cromatográfica.
46.2.10.1 Columna que satisfaga las condiciones determinadas en 41.4.2 de los Métodos Oficiales de Análisis de Aceites y Grasas.
46.2.10.2 Columna de vidrio con diámetro interior de 4 mm y una longitud aproximada de 2 mm. Rellenada con Chromosorb G, W o Q (80-100 mallas), conteniendo de 2,5 a 5 por 100 de un poliéster, siendo recomendable cualquiera de los tres siguientes: dietilenglicolsuccinato (DEGS), etilenglicolsuccinato o adipato (EGS o EGA), polietilenglícoladipato (PEGA).
Antes de emplear una columna nueva en la resolución de problemas analíticos, debe ser acondicionada eliminando todos aquellos productos volátiles que perturbarían la marcha de la cromatografía. Para ello, se monta en el cromatógrafo, sin conectarla al detector, y se calienta el horno a unos 10° por encima de la temperatura máxima a que vaya a ser utilizada la columna en trabajos posteriores; haciendo pasar, al mismo tiempo, una corriente de nitrógeno de 30 a 40 ml/min, que se mantiene durante veinticuatro horas, como mínimo. La columna será apta para su utilización si, una vez conectada al detector y en funcionamiento normal, la línea base dibujada por el registrador acusa la estabilidad del sistema.
46.3 Reactivos.
46.3.1 Metanol absoluto (99,8 por 100).
46.3.2 Hidróxido potásico, en lentejas.
46.3.3 Éter de petróleo o hexano.‒Éter de petróleo (p. e. 40°-60 °C), cuyo contenido en benceno no sea superior a 0,1 por 100, hexano normal, que cumpla las mismas especificaciones del éter de petróleo.
46.3.4 Heptano normal, con una riqueza mínima en heptano normal, determinado por cromatografía gaseosa, del 99 por 100.
46.3.5 Disolución 2 N de hidróxido potásico en metanol.‒Disolver 11,2 g de hidróxido potásico en 100 ml de metanol.
46.3.6 Ésteres metílicos de pureza adecuada para su utilización como patrones en cromatografía gaseosa.‒Se dispondrá de los ésteres metílicos de los ácidos mencionados a continuación, debiendo tener una pureza mínima de 99 por 100, determinada por cromatografía gaseosa:
Ácido butanoico (botirico).
Ácido pentanoico (valeriánico).
Ácido hexanoico (caproico).
Ácido octanoico (caprílico).
Ácido decanoico (cáprico).
Ácido dodecanoico (láurico).
Ácido tetradecanoico (mirístico).
Ácido hexadecanoico (palmítico).
Ácido octadecanoico (esteárico).
Ácido 9-octadecanoico (oleico).
Ácido 9,12 octadecadienoico (linoleico).
Ácido eicosanoico (aráquico).
46.3.7 Solución de referencia I.‒En un matraz aforado de 50 ml, se pesa, con exactitud de ± 0,1 mg, 1 g de pentanoato de metilo, disolviéndolo en heptano normal y completando hasta el enrase.
46.3.8 Solución de referencia II.‒En un matraz aforado de 100 ml se pesa, con exactitud de ± 0,1 mg, 200 mg de pentanoato de metilo, disolviéndolo en heptano normal y completando hasta el enrase.
46.4 Procedimiento.
46.4.1 Preparación de los ésteres metílicos.
En un matraz de fondo redondo de 50 ml, pesar, con exactitud de ± 0,1 mg, 1 g de grasa. Añadir 10 ml de éter de petróleo o hexano y agitar suavemente hasta disolución de la grasa.
En el caso de que se quiera efectuar una determinación cuantitativa de los ácidos butírico y caproico en la muestra, agregar a la disolución en éter de petróleo de la grasa 1 ml, exactamente medido, de la solución de referencia más adecuada; para muestras conteniendo de 1-4 por 100 de ácido butírico se utilizará la solución de referencia I; para muestras conteniendo menos de 1 por 100 de ácido butírico, se utilizará la solución de referencia II.
Si se desea efectuar solamente un análisis completo de la fracción de ácidos grasos, para lo que se aplica el método de normalización interna, no será necesario el empleo de solución de referencia.
A la solución en éter de petróleo de la muestra, adicionada o no de solución de referencia, agregar 0,5 ml de disolución 2 N de hidróxido potásico. Agitar suavemente la mezcla hasta que se ponga transparente, para lo cual son suficientes unos veinte a treinta segundos. Casi inmediatamente después de observar la clarificación de la solución, suele apreciarse un enturbiamiento debido a la separación de glicerol, que se sedimenta rápidamente.
Inmediatamente después de terminada la reacción y observada la sedimentación, tomar la cantidad necesaria con la jeringa e inyectar en el cromatógrafo, una demora en la inyección de los ésteres metílicos daría lugar a la formación de jabones, con error en la determinación.
46.4.2 Determinación cromatográfica.
46.4.2.1 Condiciones de trabajo.
Temperatura de la columna: Temperatura programada de 60 °C a 180 °C, con una velocidad de 4 °C/min.
Temperatura del inyector: 200 °C.
Temperatura del detector: 200 °C
Gas portador: Nitrógeno (o helio), con un flujo de 60 ml/min.
Flujo de hidrógeno y aire para la alimentación del detector: Los flujos dependerán del tipo de detector utilizado, debiendo determinarse previamente para optimizar la respuesta.
El registro obtenido del cromatograma debe satisfacer las condiciones que se indican a continuación; caso contrario, se repite la inyección modificando la cantidad inyectada o la sensibilidad de trabajo hasta obtener un cromatograma satisfactorio.
Los requisitos exigibles son los siguientes:
a) El área total descrita en el registro, referida a la sensibilidad máxima utilizada en el curso de la operación, debe ser de un orden aproximado de 2.000 mm2, con una velocidad del papel en el registrador de 5 mm/min. De esta forma, los componentes presentes en una cuantía del 0,1 por 100 deben dar un pico, como mínimo, de 2 mm2, siendo, por tanto, perfectamente reconocibles.
b) Con el fin de conseguir que todos los picos caigan dentro del papel registrador, se utilizará, en cada caso, la atenuación de sensibilidad que sea necesaria, cuidando que el pico de mayor intensidad no sea atenuado más de ocho veces.
Una vez conseguido un registro satisfactorio, y habiendo alcanzado nuevamente la pluma la línea base, se interrumpe el funcionamiento del registrador y se retira el papel con el registro para la identificación de los picos y/o cálculos cuantitativos.
46.4.2.2 Identificación de los picos.‒Se seguirán los criterios establecidos en el método número 41 de los Métodos Oficiales de Análisis de Aceites y Grasas.
46.4.2.3 Determinaciones cuantitativas.‒La determinación cuantitativa se basa en el principio de que los pesos de cada uno de los componentes separados en la mezcla son proporcionales a las áreas comprendidas dentro de los triángulos dibujados debajo de cada pico. El área de cada triángulo se obtiene trazando rectas tangentes a las líneas dibujadas en el registro, prolongándolas hasta su intersección con la línea base y multiplicando la altura del triángulo por la mitad de la base. En el caso de haber trabajado con atenuaciones diferentes para cada pico, se referirán todas las medidas a una misma sensibilidad del registrador, multiplicando la altura por el factor de atenuación correspondiente en cada caso, y el valor de la altura así corregida por la mitad de la base.
46.4.3 Determinación del contenido de los ácidos butírico y caproico en la materia grasa.‒Esta determinación se realiza por el método del patrón interno, siendo el patrón elegido el pentanoato de metilo.
46.4.3.1 Preparación de la mezcla de calibración.‒Con una exactitud de ± 0,1 mg y en un matraz aforado de 50 ml, pesar unos 100 mg de cada uno de los siguientes patrones: butanoato de metilo, pentanoato de metilo y caproato de metilo. Se disuelve la mezcla de heptano normal, y se diluye completando hasta el enrase.
Inyectar la cantidad necesaria de la solución anterior, normalmente 0,2-0,4 μl, para que, trabajando a la sensibilidad media del aparato, se consiga situar los máximos de los picos en una posición del 70-80 por 100 del recorrido total de la pluma del registrador. Los tres picos deberán registrarse a la misma sensibilidad. Si fuese necesario, se diluirá la solución anterior con heptano normal en la relación necesaria para poder ajustarse a las prescripciones fijadas. Efectuar, cuando menos, tres determinaciones consecutivas, que no deben discrepar entre sí más del 1 por 100.
46.4.4 Análisis cuantitativo de la totalidad de los componentes de la fracción de ácidos grasos, comprendiendo del C4 al C20 y C18:3.
46.4.4.1 Preparación de la mezcla de calibración.‒Determinar previamente el factor de corrección para cada ácido componente de la mezcla, referido a uno cualquiera de ellos, que se toma como patrón, eligiéndose normalmente para este fin el ácido palmítico, y debiendo tener la mezcla de calibración una composición análoga a la de la mezcla problema.
Para ello, si no se conoce previamente el orden de composición del problema, se realizará una determinación cromatográfica de orientación, realizándose en el registro la cuantificación de los componentes suponiendo el mismo factor de respuesta para todos ellos, efectuando un reparto proporcional entre las áreas medidas.
En un matraz aforado de 50 ml, pesar, con una exactitud de ± 0,1 mg, cantidades de los ésteres metílicos patrones que se indican a continuación proporcionales a las cifras de composición encontradas en el análisis de orientación anteriormente aludido, o previstas con anterioridad para la muestra. Los patrones que deben pesarse son los siguientes: butanoato de metilo, hexanoato de metilo, octanoato de metilo, decanoato de metilo, dodecanoato de metilo, oleato de metilo, linoleato de metilo y elcosanoato de metilo. Se disuelve la mezcla de heptano normal, agregando la cantidad adecuada de disolvente en relación al peso total de ésteres metílicos que se hayan pesado; para unos 500 mg en total, se deben emplear, como orientación, unos 50 ml de heptano. A continuación inyectar 0,2-0,4 μl, para que, trabajando a la sensibilidad media del aparato, se consiga situar el máximo del pico correspondiente al componente mayoritario, en una posición del 70-80 por 100 del recorrido total de la pluma del registrador. Todos los picos deben registrarse a la misma sensibilidad, lo cual suele ser perfectamente factible en la grasa de leche; en aquellos casos en que la relación entre el pico mayoritario y el pico minoritario no permita registrar este último con las dimensiones adecuadas para efectuar una cuantificación correcta de su área, se podrá efectuar el cambio necesario en la atenuación del registro, procurando que ésta no sobrepase la relación de 4:1. Si fuese necesario, se diluirá la solución con heptano normal en la relación necesaria para poder ajustarse a las prescripciones finadas. Efectuar, cuando menos, tres determinaciones consecutivas, que no deben discrepar entre sí más del 1 por 100.
46.5 Cálculos.
46.5.1 Cálculo de los factores de corrección para los ácidos butírico y caproico.‒Se determinan las áreas de los tres picos, siguiendo las normas que se contienen en el apartado 6.4, se calculan los dos factores correspondientes al C4 y C6 con la fórmula siguiente:

Siendo:
fx = factor de corrección del ácido.
x = cantidad pesada del ácido.
Ap = área medida en el registro para el patrón de pentanoato.
AX = área medida en el registro para el ácido x.
P' = peso del patrón pentanoato.
46.5.2 Cálculo del contenido de ácidos.‒Los contenidos de ácido butírico y ácido caproico en la muestra de grasa se calculan por la fórmula siguiente:

Siendo:
fx = factor de corrección determinado para cada ácido, según se indica en el párrafo anterior.
Ax = área medida en el registro para el ácido.
P = peso del patrón interno (pentanoato).
Ap = área medida en el registro para el patrón interno.
M = peso de la muestra de grasa.
46.5.3 Cálculo de los factores de corrección.‒Una vez determinadas las áreas de todos los picos, siguiendo las normas que se contienen en el apartado 46.4.2.2, se calcula el factor de cada ácido, referido al ácido palmítico tomado como unidad, utilizando la fórmula que se incluye en el apartado 46.5.1, sustituyendo el área Ap y el peso P del compuesto patrón por los valores correspondientes al palmitato de metilo.
46.5.4 Cálculo de composición de la fracción de ácidos grasos.‒Se calcularán las áreas corregidas de cada uno de los componentes de la fracción multiplicando el área medida en el registro por el factor de corrección determinado según se indica en el apartado anterior. El contenido de cada componente vendrá dado por la expresión:

Siendo:
fx = factor de corrección del componente x.
Ax = área medida en el registro para el componente x.
Σ (fx Ax) = suma de todas las áreas corregidas correspondientes a los componentes de la fracción.
46.6 Referencias.
1. Instituto Nacional de Racionalización y Normalización del Trabajo. Una Norma Española. 55.118.
47. FÓSFORO
47.1 Principio.
Incineración de la muestra en presencia de óxido de cinc, seguida de la medida colorimétrica del fósforo como azul de molibdeno.
Aplicable a aceites vegetales brutos, desgomados y refinados.
47.2 Material y aparatos.
47.2.1 Crisoles de porcelana de 50 ml de capacidad.
47.2.2 Vidrios de reloj.
47.2.3 Placa de calefacción eléctrica, con regulador de temperatura.
47.2.4 Horno de mufla.
47.2.5 Embudo de vidrio de vástago corto de 50 mm de diámetro.
47.2.6 Papel de filtro de 90 mm de diámetro, Albet 242 o similar.
47.2.7 Frasco lavador de un litro de capacidad con cuello protegido del calor.
47.2.8 Matraces aforados de 50, 100, 250 y 500 ml de capacidad provistos de tapones de vidrio.
47.2.9 Pipetas de 2 5, 10 y 25 ml de capacidad.
47.2.10 Pipetas de 10 ml de capacidad, divididas en décimas de mililitro.
47.2.11 Espectrofotómetro para medición en el visible.
47.2.12 Cubetas espectrofotométricas de 10 mm de paso.
47.3 Reactivos.
47.3.1 Ácido clorhídrico (d = 1,19).
47.3.2 Oxido de cinc.
47.3.3 Hidróxido potásico.
47.3.4 Ácido sulfúrico (d = 1,84),
47.3.5 Molibdato sádico.
47.3.6 Sulfato de hidracina.
47.3.7 Fosfato ácido monopotásico, desecado a 103 ± 2 °C durante dos horas antes de usarlo.
47.3.8 Disolución de molibdato sódico.‒Añadir con precaución 140 ml de ácido sulfúrico a 300 ml de agua destilada. Dejar enfriar la temperatura ambiente, añadir 12,5 g de molibdato sódico y disolver totalmente. Llevar a un matraz aforado de 500 ml, diluir con agua destilada hasta el enrase, homogeneizar y dejar reposar durante veinticuatro horas, por lo menos, antes de usarla.
47.3.9 Disolución de sulfato de hidracina al 0,015 por 100.‒Disolver en un matraz aforado 0,15 g de sulfato de hidracina en un litro de agua destilada.
47.3.10 Disolución de hidróxido potásico al 50 por 100 (m/m). Disolver 50 g de hidróxido potásico en 50 ml de agua destilada.
47.3.11 Disolución de fosfato ácido monopotásico.‒Disolver 1,0967 g de fosfato monopotásico seco en agua destilada. Diluir a 250 ml en un matraz aforado y agitar. Esta disolución contiene 1 mg de fósforo por mililitro. A continuación verter, con una pipeta, 5 ml de la disolución anterior en un matraz aforado de 500 ml y enrasar con agua destilada. Esta disolución contiene 0,01 mg de fósforo por mililitro.
47.4 Procedimiento.
47.4.1 Construcción de la curva patrón.‒Tomar, mediante pipeta, 1, 2, 4, 6, 8 y 10 ml de la disolución 47.3.11, e introducir en matraces aforados de 50 ml. Completar el volumen a 10 ml con agua destilada, utilizando la pipeta 47.2.10, continuando como se describe en 47.4.3 a partir de la adición de sulfato de hidracina. Las alícuotas tomadas de la disolución contienen 0,01, 0,02, 0,04 0,06, 0,08 y 0,10 mg de fósforo. A continuación representar gráficamente los valores de absorbancia frente a los contenidos en fósforo, expresado en mg.
47.4.2 Preparación de la muestra.‒Pesar, con precisión de 1 mg, de 3 a 3,2 g de muestra en un crisol de porcelana y añadir 0,5 g de óxido de cinc. Calentar lentamente en la placa eléctrica hasta que la muestra se espese y entonces aumentar gradualmente la calefacción hasta que la masa esté completamente carbonizada. Colocar el crisol en el horno de mufla a una temperatura de 550-600 ºC, manteniéndolo durante dos horas. Una vez transcurrido ese tiempo, dejar enfriar a la temperatura ambiente.
Añadir a las cenizas 5 ml de agua destilada y 5 ml de ácido clorhídrico. Cubrir el crisol con un vidrio de reloj y calentar suavemente a ebullición durante cinco minutos. Filtrar la disolución recogiendo el filtrado en un matraz aforado de 100 ml. Lavar la parte interior del vidrio de reloj y las paredes del crisol con 5 ml de agua destilada caliente, usando un frasco lavador con chorro de agua finísimo. Lavar el crisol y el papel de filtro con cuatro porciones de 5 ml de agua destilada caliente. Enfriar la disolución a temperatura ambiente y neutralizar hasta débil turbidez, adicionando unas gotas de potasa al 50 por 100. Añadir ácido clorhídrico gota a gota hasta que el precipitado de óxido de cinc se disuelva y entonces añadir dos gotas más. Diluir hasta 100 ml con agua destilada y agitar.
47.4.3 Determinación.
Pasar, mediante pipeta, 10 ml de esta disolución a un matraz aforado de 50 ml. Añadir 8 ml de la disolución de sulfato de hidracina y a continuación 2 ml de la disolución de molibdato sódico. Tapar e invertir el matraz aforado dos o tres veces, quitar el tapón y calentar durante 10 ± 0,5 minutos en baño de agua hirviendo. Transcurrido este tiempo retirar del baño, enfriar a 25 ± 5 ºC en baño de agua, diluir hasta el enrase con agua destilada y mezclar perfectamente. Llenar una cubeta del espectrofotómetro y medir la absorbancia a 650 nm utilizando como referencia agua destilada.
Realizar un ensayo en blanco como se ha descrito, pero sin añadir aceite.
47.5 Cálculos.
Calcular el contenido en fósforo, expresado en tanto por ciento, mediante la siguiente fórmula:

Siendo:
M = contenido en fósforo, en mg, de la alícuota.
B = contenido en fósforo, en mg, del ensayo en blanco.
P = peso, en g, de la muestra.
V -= volumen, en ml, tomados en 47.4.3.
% en fosfátidos = % fósforo X F, donde F = factor de conversión de fósforo en fosfátidos (ver 47.6.2).
47.6 Observaciones.
47.6.1 No debe demorarse la lectura espectrofotométrica una vez conseguido el desarrollo del color.
47.6.2 En el caso del aceite de soja el valor de V es prácticamente igual a 30.
47.7 Referencias.
1. Instituto Nacional de Racionalización y Normalización del Trabajo. Una Norma Española. 55.108-73.
48. GRASA NEUTRA
48.1 Principio.
Retención de los ácidos grasos libres y sustancias no grasas por cromatografía en columna de alúmina. Los productos no retenidos en la columna se pesan, obteniéndose por diferencia a 100 los ácidos grasos libres e impurezas retenidas en la columna.
No es aplicable con carácter general a los aceites de orujos.
48.2 Material y aparatos.
48.2.1 Equipo para cromatografía en columna, compuesto de:
48.2.1.1 Columna de vidrio, provista de una placa de vidrio porosa número 2, correspondiente a un diámetro medio de poro de 40 a 90 μ (fig. 48.1).
48.2.1.2 Depósito de disolvente para la alimentación de la columna (fig. 48.2).
48.2.1.3 Soporte para el manejo del frasco de pesada (figura 48.3).
48.2.1.4 Frasco de pesada de unos 20 ml de capacidad (figura 48.4).
48.2.1.5 Tubo de prolongación para el vaciado del frasco de pesada en la columna (fig. 48.5).
48.2.1.6 Matraz redondo, fondo plano, de vidrio Pyrex o similar, de 250 ml de capacidad y boca esmerilada normalizada 29/32.
48.2.2 Ampolla de decantación de 125 ml de capacidad, con llave de vidrio.
48.2.3 Frasco lavador de unos 125 ml de capacidad.
48.2.4 Estufa de vacío que permita calentar hasta 110 °C como mínimo y regulación de ± 1 °C. La temperatura será además uniforme en todo el espacio interior, tolerándose diferencias que no superen 1 °C entre posiciones extremas.
48.2.5 Horno de mufla capaz de alcanzar una temperatura de 800 °C y regulación de ± 5 °C.
48.2.6 Desecador conteniendo un agente desecante apropiado.
48.2.7 Evaporador rotatorio.
48.2.8 Cápsulas de porcelana, fondo redondo de 50 mm 0 y 20 mm de altura.
48.2.9 Pesa sustancias de vidrio, de 60 mm ∅ interior y 40 de altura.
48.2.10 Tubo de cromatografía, de 10 cm de longitud y 1,5 centímetros de diámetro interior, provisto de llave esmerilada, con orificio de paso de 1,5 a 2 mm y placa filtrante de vidrio porosidad número 2, correspondiente a un tamaño medio de poro de 40 a 90 μ.
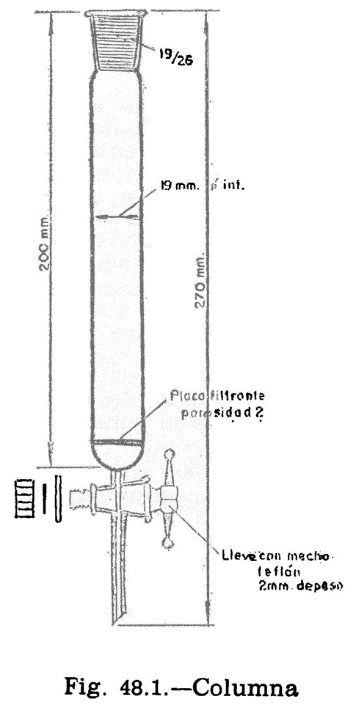
48.3 Reactivos.
48.3.1 Éter etílico.
48.3.2 Metanol anhidro.
48.3.3 Éter etílico-metanol.‒Mezclar 975 ml de 48.3.1 y 25 ml de 48.3.2.
48.3.4 Hexano, con un contenido máximo de 0,5 por 100 de aromáticos.
48.3.5 Alúmina para cromatografía en columna de las siguientes características: pH en suspensión acuosa al 5 por 100; 7,0 ± 0,5; actividad IV según Brockmann (ver 48.6.1). Granulometría.‒El análisis granulométrico del producto debe dar un contenido inferior al 10 por 100 de partículas con un grosor igual o inferior a 63 micras e inferior al 0,1 por 100 de partículas con un grosor mayor de 250 micras, con una medida estadística para la totalidad de 120 micras ± 5 por 100.
48.3.6 p-amino-azo-benceno.
48.3.7 Rojo Sudan (Sudan II) (xileno-azo-β-naftol).
48.3.8 Amarillo Sudan (Sudan I) (benceno-azo-/3-naftol).
48.3.9 Benceno purísimo, exento de tiofeno.
48.4 Procedimiento.
48.4.1 Preparación de la columna de alúmina.
Pesar, con aproximación de 0,5 g, 20 g de alúmina e introducirlos en una ampolla de extracción de 125 ml, agregando después 50 ml del disolvente 48.3.3, con el que se llena también la columna hasta ocupar, aproximadamente, 2/3 de su capacidad. Colocar la ampolla encima de la columna, de manera que el tubo de salida coincida con el extremo superior de la columna. Abrir la llave de la ampolla dejando salir la papilla de alúmina y disolvente que caerá en el interior de la columna; al mismo tiempo, abrir la llave de la columna, regulando la salida del disolvente de forma que el nivel de líquido en el interior de la columna se mantenga prácticamente constante, procurando más bien que el nivel suba en lugar de disminuir. Cuando toda la papilla de alúmina haya pasado a la columna, arrastrar las últimas porciones adheridas a las paredes, enjuagando el recipiente con un poco del mismo disolvente. Mantener abierta la llave de la columna, dejando salir lentamente el disolvente hasta que el nivel se haya situado a la altura del relleno de alúmina. Agregar hexano a la columna hasta situar el nivel a 1 cm, aproximadamente, del extremo inferior del cono esmerilado.
48.4.2 Preparación de la muestra de grasa.
Introducir el frasco de muestra en una estufa de aire regulada a la temperatura necesaria para conseguir la fusión completa de la muestra, lo que se consigue normalmente calentando de 30 a 40 °C. Conseguida la fusión, si se observa la presencia de sedimentos o materia en suspensión no fundible, agitar fuertemente el frasco hasta conseguir la homogeneización de su contenido y, una vez conseguido, efectuar inmediatamente la pesada de la muestra en el frasco de pesada (fig. 4).
48.4.3 Fraccionamiento cromatográfico.
Pesar, con aproximación de 0,2 mg, 5 g de muestra en el frasco de pesada (fig. 4), utilizando para ello el soporte adecuado (fig. 3). Una vez efectuada la pesada, separar del soporte el frasco con el aceite, ajustando el tubo de prolongación (fig. 5) al cono situado en la parte inferior del frasco.
Ajustar el frasco, con el tubo de prolongación, a la boca superior de la columna cromatográfica y verter en el interior del frasco con el aceite el disolvente 48.3.3, utilizando el frasco lavador y dirigiendo el churro a las paredes con el fin de lavar y arrastrar hacia el fondo el aceite que haya podido quedar adherido a la superficie; con el mismo chorro se mezclará todo el contenido para conseguir la homogeneización de la disolución de aceite.
Continuar agregando disolvente hasta que el nivel quede ligeramente por debajo del extremo superior del sifón; dirigiendo el chorro a la pared lateral superior, seguir agregando cuidadosamente hasta que el nivel líquido sobrepase ligeramente la altura del sifón y comience a caer por el tubo vertiendo en la columna cromatográfica.
Ajustar el depósito del disolvente (fig. 2) a la boca del frasco, cargándose con 125 ml de disolvente, colocar el matraz de 250 ml, previamente desecado a 105 °C y pesado, a la salida de la columna, con el fin de recoger el líquido que fluye de la misma. Abrir la llave del depósito y la de la columna, regulándose ambas de forma que el líquido fluya en el matraz a una velocidad de 4-5 ml por minuto, manteniéndose el nivel en el frasco por encima del extremo superior del sifón. Si el disolvente no fluye al frasco por la llave del depósito, tapar el tubo hasta que comience el flujo; quitar el tapón y continuar la operación normalmente. El desarrollo quedará terminado cuando haya pasado la totalidad del líquido y no caiga disolvente en el matraz.
La disolución recogida en el matraz se concentra en un evaporador rotatorio con vacío, calentando a la temperatura conveniente para evitar una ebullición tumultuosa que pudiera provocar proyecciones violentas del líquido.
Una vez eliminado el disolvente, llevar el matraz a una estufa de vacío para eliminar los residuos volátiles que queden en el aceite, manteniéndose el matraz en la estufa durante una hora. Sacar y pasar a un desecador, donde se deja enfriar, pesando a continuación. Repetir este tratamiento en operaciones sucesivas, hasta conseguir peso constante, con precisión del mg.
48.5 Cálculos.

Siendo:
P = peso del matraz con el aceite.
Po = peso del matraz vacío.
M = peso de la muestra en gramos.
% pérdida cromatográfica = 100 ‒ % de aceite neutro.
48.6 Observaciones.
48.6.1 La determinación de la actividad de la alúmina según Brockmann se realiza en la forma siguiente:
La alúmina que se ha de ensayar se introduce en la columna cromatográfica 48 2.10, procurando conseguir una distribución homogénea, formando una capa de 5 cm de altura y colocando en la parte superior un disco de papel de filtro. Preparar una disolución mezclando 2 ml de benceno, 8 ml de hexano, 2 mg de rojo Sundan, 2 mg de p-amino-azobenceno y 2 mg de amarillo Sudan. Verter la disolución de colorantes en la columna, abrir la llave y dejar pasar la disolución hasta que el nivel líquido quede justamente a la altura de la alúmina. Verter a continuación el líquido de desarrollo constituido por una mezcla de benceno-hexano en la proporción 4:1 en volumen, del que se hacen pasar por la columna 20 ml. La velocidad de salida se regula con la llave, manteniendo un flujo de 0,5 a 1 ml por minuto, recogiendo el líquido en un matraz cónico de boca estrecha
Terminado el desarrollo, con una alúmina de grado IV que es la apropiada, el amarillo Sudan habrá sido eliminado totalmente de la columna, quedando retenidos los otros dos que aparecerán en dos bandas: Una inferior roja, correspondiente al rojo Sudan y otra superior amarilla correspondiente al aminoazo-benceno. Una situación distinta de estos colorantes deberá interpretarse como un grado de actividad mayor o menor del producto, requiriendo el ajuste de su contenido en agua de la forma conveniente.
48.6.2 Preparación de la alúmina.
Partiendo de una alúmina de actividad superior a IV, se adicionarán cantidades crecientes de agua hasta conseguir en el ensayo de actividad, realizado como se indica en el apartado 48.6.1, el grado IV necesario.
Efectuado esto, si se dispone de una partida relativamente importante de alúmina, es conveniente determinar su humedad, siguiendo la metódica que se describe en 4.6.3, conocido este dato, en operaciones sucesivas, bastará con determinar la humedad de la alúmina, calculando la cantidad de agua que hay que añadir para obtener su producto de actividad IV.
Para incorporar a la alúmina una determinada cantidad de agua, se procederá de la forma siguiente: Introducir la alúmina pesada en un matraz o frasco con tapón esmerilado con una capacidad aproximadamente el doble del volumen de alúmina. Agregar el peso calculado de agua destilada, tapar el recipiente y agitar hasta conseguir la homogeneización del producto. Mantener el frasco cerrado durante 48 horas como mínimo, agitándolo durante este tiempo unas 10 o 12 veces por lo menos. Después de este tratamiento la alúmina debe estar lista para su utilización.
48.6.3 Determinación de la humedad de la alúmina.
En una capsulita de porcelana pesar, con precisión de 1 miligramo, 1 g de alúmina. Introducir en la mufla del horno y mantenerla 2 horas a 600 °C. Dejar enfriar parcialmente e introducir en un pesasustancias de dimensiones adecuadas; tapar y llevar a un desecador donde se mantiene hasta adquirir la temperatura ambiente. Pesar la totalidad, sin destapar el pesa-sustancias.
Previamente la cápsula vacía se habrá calentado en el horno a 600 °C, durante media hora como mínimo; y el pesasustancias, en una estufa de aire a 105 °C, durante una hora, dejándolos enfriar en el desecador. Las piezas así desecadas se pesarán vacías.

Siendo:
P1 = peso de la cápsula y el pesasustancias con la alúmina.
Po = peso de la cápsula y el pesasustancias con la alúmina, una vez desecada.
P = peso exacto de la muestra de alúmina.
48.7 Referencias.
1. Instituto de Racionalización y Normalización del Trabajo. Una Norma Española, 55.105.
1. PREPARACIÓN DE LA MUESTRA PARA EL ANÁLISIS
1.1 Principio.
Las operaciones descritas a continuación tienen por finalidad conseguir una muestra para el análisis lo más homogénea posible. Por ello, toda simplificación o tratamiento insuficiente en esta operación puede conducir a unos resultados que no sean representativos.
1.2 Material y aparatos.
1.2.1 Cuchillo.
1.2.2 Trituradoras eléctricas de distinto grado de finura en el picado.
1.2.3 Frascos de vidrio de 250 ml de capacidad, de color topacio, boca ancha y tapón esmerilado.
1.2.4 Cápsulas de porcelana de 20 cm de diámetro.
1.3 Procedimiento.
1.3.1 Tomar una muestra representativa de 200 g.
1.3.2 Quitar la piel si la tuviere (embutidos, etc.).
1.3.3 Partirla con cuchillo en rodajas o trozos de 0,5-1 cm.
1.3.4 Cortar los trozos en pequeños cubos.
1.3.5 Pasarlos varias veces por trituradora hasta conseguir una mezcla homogénea.
1.3.6 La muestra, bien homogeneizada debe guardarse inmediatamente en los frascos, limpios y secos, de forma que queden llenos, para prevenir pérdidas de humedad.
1.3.7 Conservarlos en refrigeración de forma que evite su deterioro y cualquier cambio en su composición.
1.3.8 Tomar las muestras para las diferentes determinaciones a ser posible dentro de las 24 horas siguientes.
2. ALMIDÓN
(Método cualitativo)
2.1 Principio.
El almidón reacciona con el iodo dando coloración azul.
2.2 Material y aparatos.
2.2.1 Erlenmeyer de 150 ml de capacidad.
2.2.2 Frasco cuentagotas.
2.2.3 Pipeta de 10 ml de capacidad.
2.3 Reactivos.
2.3.1 Solución iodo-iodurada:
Mezclar 1 g de iodo y 2 g de ioduro potásico en agua destilada hasta 200 ml.
Mantener la solución en el frasco cuentagotas.
2.4 Procedimiento.
2.4.1 Preparación de la muestra:
Como en el método 1.
2.4.2 Valoración:
Introducir 10 g de la muestra preparada como 2.4.1 en un Erlenmeyer de 150 ml. Añadir 40 ml de agua destilada. Hervir durante 5 minutos. Transcurrido este tiempo enfriar exteriormente el matraz en corriente de agua fría.
Con pipeta de 10 ml atravesar la capa grasa superior, tomando 10 ml del líquido inferior, transvasándoles a un tubo de ensayo. Añadir 5 gotas de la solución 2.3.1.
2.5 Interpretación de resultados.
En presencia de almidón aparecerá una coloración azul-negra.
3. ALMIDÓN
(Método cuantitativo)
3.1 Principio.
Extracción de azúcares simples con etanol caliente 80 por 100, permaneciendo el almidón.‒El residuo de almidón se solubiliza con ácido perclórico diluido, y medida a 630 nm del color desarrollado al calentarlo con el reactivo antrona-sulfúrico.
3.2 Material y aparatos.
3.2.1 Balanza analítica.
3.2.2 Matraces aforados de 100 ml y 200 ml.
3.2.3 Tubos de centrífuga, cónicos, de 100 ml.
3.2.4 Pipetas graduadas de 10 ml, 25 ml, 5 ml y 2 ml.
3.2.5 Centrífuga de 2.500 r.p.m.
3.2.6 Baño de agua.
3.2.7 Baño de agua termostatable hasta 25 °C.
3.2.8 Probeta graduada de 25 ml.
3.3 Reactivos.
3.3.1 Disolución de ácido sulfúrico-antrona.‒Disolver 0,2 g de antrona en 100 ml de H2SO4. El reactivo sirve para 3-4 días conservándolo a 0 °C.
3.3.2 Glucosa patrón.‒Disolver 0,1 g de glucosa anhídrica en 100 ml de agua.
3.3.3 Ácido perclórico al 52 por 100.
3.3.4 Etanol al 80 por 100.
3.3.5 Etanol puro.
3.3.6 Éter de petróleo.
3.3.7 Disolución etanol-éter de petróleo (1/3) (v/v).
3.4 Procedimiento.
3.4.1 Extracción de azúcar y de grasa.‒Pesar 2 g de carne triturada preparada como en el método 1 en un tubo centrífugo cónico 100 ml. Añadir 25 ml de disolución etanol-éter de petróleo (1-3), tapar con tapón, agitar vigorosamente y centrifugar a 2.500 r.p.m. durante 5 minutos. Decantar y dejar a un lado la disolución etanol-éter de petróleo. Añadir 10 ml de etanol caliente al 80 por 100, agitar y centrifugar a 2.500 r.p.m. durante 5 minutos. Dejar a un lado la disolución alcohólica y repetir la extracción alcohólica con etanol caliente.
3.4.2 Extracción de almidón.‒Añadir 5 ml de agua al residuo y remover. Añadir 6,5 ml de disolución de ácido perclórico diluido (52 por 100); remover o agitar durante 5 minutos. Dejar reposar durante 15 minutos. Añadir 20 ml de agua y centrifugar durante 5 minutos. Verter la disolución de almidón en un frasco volumétrico de 100 ml. Añadir 6,5 ml de disolución de ácido perclórico (52 por 100) y remover. Dejar reposar durante treinta minutos. Remover y lavar el contenido entero del tubo en el frasco volumétrico que contiene el primer extracto. Llevar a volumen y filtrar por papel filtro (3.2.9).
3.4.3 Determinación de almidón.‒Diluir 5 ml de disolución de almidón filtrada en 200 ml con agua destilada. Pipetear 5 ml de dicha disolución en un tubo, enfriar en baño de María y añadir 10 ml de antrona reactivo (3.3.1) Mezclar completamente y calentar durante 7,5 minutos a 100 °C. Quitar el tubo del baño, enfriar con rapidez a 25 °C y determinar la absorbancia a 630 mm. El color permanece fijo durante treinta minutos.
3.5 Cálculos.
3.5.1 Curva patrón de glucosa.‒Diluir 1, 2, 5 y 10 ml de 3.3.2 hasta 100 ml con agua destilada. A partir de las lecturas obtenidas, dibujar la curva patrón.
3.5.2 Contenido en almidón de la muestra:
% glucosa = 0,04 • μg de glucosa leídos en la curva patrón.
% almidón = 1,06 • % glucosa.
3.6 Observaciones.
3.6.1 Teniendo en cuenta que el carragenato se determina como almidón, se deberá deducir del valor obtenido de almidón el correspondiente de carragenato.
3.6.2 En los productos que declaren contener carragenato y hasta que no exista técnica oficial para su determinación cuantitativa, se permitirá una tolerancia del 1,4 por 100 en el valor obtenido de almidón.
3.7 Bibliografía
1. W. Glover, H. Kirschenbaum y A. Caldwell (Departamento de Consumo y Comercialización, mlnisterio de Agricultura de los Estados Unidos, Laboratorios de Inspección de Carne, Nueva York, N.Y. 10011).
4. CONSERVADORES
(Por cromatografía en capa fina)
4.1 Principio.
Extracción de los conservadores por medio de una mezcla de éter y éter de petróleo y posterior identificación por cromatografía en capa fina.
4.2 Material y aparatos.
4.2.1 Homogeneizador.
4.2.2 Matraces.
4.2.3 Erlenmeyer de 25 y 250 ml.
4.2.4 Refrigerantes de reflujo.
4.2.5 Filtro Büchner y papel de filtro de filtración rápida.
4.2.6 Embudos y papel de filtro plegados.
4.2.7 Ampolla de decantación de 100 y 250 ml.
4.2.8 Cápsulas de porcelana de fondo plano.
4.2.9 Tubos pequeños de cristal con tapón.
4.2.10 Material para cromatografía en capa fina:
‒ Cubeta.
‒ Micropipetas.
‒ Secador.
‒ Fuente de ultravioleta.
‒ Placas de poliamida (20 x 20) con soporte de aluminio y revelador de fluorescencia incorporado tipo F-254 Merck, o equivalente.
4.3 Reactivos.
4.3.1 Aoluxión acuosa de ácido sulfúrico al 10 por 100 (v/v).
4.3.2. Reactivo de Carrez (para defecación).
4.3.2.1 Solución acuosa de ferrocianuro potásico al 15 por 100 (p/v).
4.3.2.2 Solución acuosa de acetato de cinc al 30 por 100 (p/v).
4.3.3 Cloruro sódico.
4.3.4 Mezcla de éter y éter de petróleo (1/1).
4.3.5 Solución acuosa saturada de cloruro sódico.
4.3.6 Sulfato de sodio anhidro.
4.3.7 Soluciones testigo conteniendo 3 g/l en la mezcla a partes iguales de éter y éter de petróleo de los siguientes conservadores:
4.3.7.1 Ácido salicílico.
4.3.7.2 Ácido benzoico.
4.3.7.3 Ácido clorobenzoico.
4.3.7.4 Ácido parahidroxibenzoico.
4.3.7.5 Éster etílico del ácido parahidroxibenzoico.
4.3.7.6 Ácido ascórbico.
4.3.8 Eluyentes:
4.3.8.1 Mezcla de n-pentano, n-hexano, ácido acético (10, 10, 3).
4.3.8.2 Mezcla de benceno, acetato de etilo, ácido acético (85, 10, 5).
4.4 Procedimiento.
4.4.1 Extracción de los conservadores.
Tomar aproximadamente 50 g de la muestra preparada como en el método 1 y transvasarla a un matraz Erlenmeyer de 250 ml.
Añadir 15 ml de solución de ácido sulfúrico al 10 por 100 diluidos en 60 ml de agua hirviendo. Añadir 10 ml de solución de ferrocianuro potásico y agitar; 10 ml de solución de acetato de cinc y agitar.
Adaptar el refrigerante de reflujo. Someter la mezcla durante treinta minutos a ebullición bajo un ligero reflujo. Filtrar rápidamente en caliente sobre un disco de papel de filtro mojado colocado sobre un embudo. El filtrado queda generalmente turbio. Añadir 2 ml de ferrocianuro potásico y agitar 2 ml de solución de acetato de cinc y agitar. Añadir 20 g, aproximadamente, de cloruro sódico. Filtrar en caliente (aproximadamente a 80 °C) sobre filtro plegado, mojado, de forma que se retengan las grasas. Enfriar el filtrado. Trasvasar el filtrado a un embudo de decantación de 250 ml. Extraer los conservadores de la fase acuosa por medio de tres lavados con 20 ml de mezcla éter y éter de petróleo agitando suavemente durante 10 minutos, aproximadamente, para evitar la formación de una emulsión. Reunir los tres extractos etéreos y eliminar la fase acuosa. Lavar dos veces la fase etérea con 20 ml de solución saturada de cloruro sódico para romper las emulsiones que eventualmente hayan podido formarse y dos veces con 20 ml de agua destilada. Transvasar la solución etérea a un matraz de 250 ml en el cual se haya puesto en el fondo un gramo de sulfato sódico anhidro. Tras dejarlo durante algunos minutos en contacto, transvasarlo a una pequeña cápsula de porcelana de fondo plano. Evaporar el disolvente a temperatura ambiente (no calentar para evitar la pérdida de ácido benzoico). Redisolver el depósito de la cápsula con algunas gotas de mezcla de éter y éter de petróleo. Lavar la cápsula dos o tres veces con el mismo disolvente para obtener un extracto de un volumen total, aproximadamente, de 0,5 ml.
4.4.2 Preparación del cromatograma.
Depositar las soluciones separadas sobre la placa de poliamida a 1 cm del borde por medio de una micropipeta (el diámetro de las manchas no debe ser mayor de 1 a 2 mm). Aplicar de la misma forma las soluciones testigo para comparar, una vez desarrollado el cromatograma, con las muestras problema, lo cual nos evita medir los Rf. Colocar la placa en la cubeta. Eluir hasta que el frente del eluyente haya recorrido los dos tercios de la placa. Sacar la placa y secar.
4.5 Interpretación de resultados.
4.5.1 El ácido salicílico ha sido escogido como sustancia de referencia, por producir fluorescencia azul de las manchas que se obtienen en los cromatogramas cuando éstos se ponen bajo luz ultravioleta, por lo cual pueden ser identificados fácil y rápidamente, bien por medida de sus Rf o bien por comparación con las manchas producidas por las soluciones testigo.
4.5.2 Rf de los conservadores.
| Ácido p-hidroxibenzoico | Éster etílico del ácido p-hidroxibenzoico | Ácido salicílico | Ácido clorobenzoico | Ácido benzoico | Ácido sórbico | |
|---|---|---|---|---|---|---|
| Eluyente 1. | 0,05 | 0,21 | 0,46 | 0,75 | 0,86 | 0,94 |
| Eluyente 2. | 0,10 | 0,56 | 0,25 | 0,54 | 0,70 | 0,82 |
5. NITRÓGENO TOTAL
5.1 Principio.
Ataque del producto por ácido sulfúrico concentrado, catalizado con sulfato de cobre y selenio, en el cual se transforma el nitrógeno orgánico en iones amonio, que en medio fuertemente básico, permite la destilación del amoníaco, que es recogido sobre ácido bórico. La posterior valoración con ácido clorhídrico permite el cálculo de la cantidad inicialmente presente de nitrógeno en la muestra.
5.2 Material y aparatos.
5.2.1 Balanza analítica.
5.2.2 Batería calefactora.
5.2.3 Probetas de 50 ml.
5.2.4 Matraces Kjeldahl de 800 ml.
5.2.5 Embudos de vástago largo.
5.2.6 Embudos de 8 cm. de diámetro.
5.2.7 Aparato de destilación.
5.2.8 Erlenmeyer de 200 ml.
5.2.9 Bureta con divisiones de 0,1 ml.
5.2.10 Frascos de 250 ml de boca ancha con rosca.
5.3 Reactivos.
5.3.1 Sulfato de cobre.
5.3.2 Sulfato potásico.
5.3.3 Ácido sulfúrico concentrado, d = 1,84.
5.3.4 Selenio en polvo.
5.3.5 Solución de hidróxido sódico al 40 por 100.
5.3.6 Solución de ácido bórico al 4 por 100.
5.3.7 Ácido clorhídrico 0,1 N.
5.3.8 Indicador.
Disolver 2 g de rojo de metilo y 1 g de azul de metileno en 1.000 ml de etanol al 95 por 100 (v/v). Este indicador vira de violeta a verde a pH 5,4. Conservarlo en frasco topacio.
5.3.9 Piedra pómez.
5.4 Procedimiento.
Pesar, con precisión de 0,1 mg, 1-3 g de la muestra, según contenido, preparada según el método 1. Llevar la muestra pesada al matraz Kjeldahl, e introducir sucesivamente unos granos de piedra pómez, 15 g de sulfato potásico, 0,5 g de sulfato de cobre, y una punta de espátula de selenio en polvo. Agregar 25 ml de ácido sulfúrico, mezclar suavemente por rotación y colocar el matraz en una batería calefactora, poniendo un embudo adecuado (5.2.5) en la boca. Calentar suavemente al principio, y cuando el conjunto adquiere una cierta decoloración aumentar la intensidad de calefacción. Agitar de vez en cuando con suavidad por rotación. Una vez que el líquido queda transparente, con una coloración azul verdosa, prolongar la ebullición al menos hora y media. Dejar enfriar hasta temperatura ambiente y añadir con precaución 100 ml de agua, disolviendo por rotación suave el sulfato potásico cristalizado.
En un Erlenmeyer de 200 ml poner 25 ml de ácido bórico al 4 por 100 y unas gotas de indicador (5.3.8). Introducir hasta el fondo en el Erlenmeyer la alargadera del aparato de destilación. Colocar el matraz en el aparato de destilación, ajustándolo bien, poniendo un poco de grasa en los esmerilados. Agregar, por el depósito superior, otros 100 ml de agua y 100 ml de disolución de hidróxido sódico al 40 por 100. Calentar suavemente hasta ebullición.
Aumentar el calentamiento recogiendo, al menos, 150 ml de destilado, o prolongarlo, hasta el momento en que se produzca una ebullición a golpes.
Retirar el Erlenmeyer, lavar la alargadera y el interior del refrigerante, recogiendo sobre el destilado las aguas de lavado. Valorar hasta la coloración original, violeta, con ácido clorhídrico 0,1 N. Efectuar una prueba en blanco, utilizando 5 ml de agua destilada en vez de la muestra, siguiendo todo el procedimiento.
5.5 Cálculo.

Porcentaje proteína total = 6,25 • porcentaje N total
Siendo:
f = factor del clorhídrico.
V1 = volumen en ml de ácido clorhídrico gastado en la valoración.
V2 = volumen en ml de ácido clorhídrico gastado en el ensayo en blanco.
p = peso en gramos de la muestra.
5.6 Referencias.
1. Norma internacional ISO R-937.
6. CENIZAS
6.1 Principio.
Adición de solución de acetato de magnesio, desecación en baño de agua o baño de arena, incineración en un horno a 550 °C y posterior determinación de la masa del residuo, teniendo en cuenta la cantidad de óxido de magnesio proveniente de la adición de la solución de acetato de magnesio utilizada en primer lugar.
6.2 Material y aparatos.
6.2.1 Cápsulas de porcelana de cuarzo o platino, con fondo plano, de aproximadamente 15 cm2 de superficie y 25 mm de altura, de paredes ligeramente inclinadas.
6.2.2 Pipeta de 1 y 2 ml de doble aforo.
6.2.3 Baño de agua o baño de arena o placa calefactora.
6.2.4 Horno de mufla provisto de termostato y capaz de alcanzar al menos 800 ºC.
6.2.5 Desecador provisto de un agente deshidratante eficaz con indicador.
6.2.6 Balanza analítica.
6.2.7 Pinzas adecuadas para el manejo de las cápsulas.
6.3 Reactivos.
6.3.1 Solución de acetato magnésico, que contenga 150 g/l. Pesar 15 g del reactivo anhidro o 25 g del tetrahidrato y llevarlos, una vez disueltos, a un matraz de 100 ml; enrasar.
6.4 Procedimiento.
Introducir la cápsula bien limpia en el horno regulado a 550 °C durante 20 minutos. Sacarla e introducirla en el desecador, permaneciendo en él 30 minutos. Pesarla con una precisión de 0,1 mg.
Introducir en la cápsula un peso P de muestra, aproximadamente 5 g, preparada según el método 1 y pesada con una precisión de 0,1 mg. Añadir 1 ml de la disolución de acetato magnésico (6.3.1) uniformemente. Colocar la cápsula en un baño de arena, o placa calefactora, hasta conseguir la carbonización de la muestra, prestando mucha atención a las posibles proyecciones. Transferir la cápsula al horno de mufla que debe quedar estabilizado a 550 °C por espacio de, 31 menos, 1 hora. Si no alcanzan el grado de blancura deseado debe sacarse la cápsula, dejarla enfriar y añadir unos mililitros (2-4) de agua destilada. Evaporar el agua en baño de arena. Introducir de nuevo la cápsula en el horno de mufla por espacio de 30 minutos y repetir las operaciones anteriores si es necesario, hasta conseguir unas cenizas blancas o ligeramente grises. Sacarlas del horno e introducirlas en el desecador durante 30 minutos.
Pesar con precisión de 0,1 mg.
Efectuar dos determinaciones sobre la misma muestra.
6.5. Cálculos.

Siendo:
Mo = masa, en gramos, de la cápsula.
M1 = masa, en gramos, de la cápsula conteniendo la muestra.
M2 = masa, en gramos, de la cápsula y el residuo después de incineración.
M3 = masa, en gramos, del óxido magnésico proveniente de la disolución de acetato magnésico añadido.
6.6 Observaciones.
6.6.1 La diferencia entre dos determinaciones sobre la misma muestra, realizadas simultáneamente o rápidamente una después de otra por el mismo analista, no debe ser superior a 0,10 g por 100 g de muestra.
6.7 Referencias.
1. Norma internacional ISO R-936.
7. FÓSFORO
7.1 Principio.
Transformación en ácido pirofosfórico, posterior hidrólisis del mismo y medida del color producido al añadirle el reactivo molibdato-vanadato.
7.2 Material y aparatos.
7.2.1 Matraces aforados de 1.000, 250 y 100 ml.
7.2.2 Pipetas de 10, 20 y 50 ml.
7.2.3 Matraces Kjeldahl de 500 u 800 ml.
7.2.4 Balanza analítica.
7.2.5 Espectrofotómetro capaz de efectuar lecturas a 436 nm.
7.3 Reactivos.
7.3.1 Solución de vanadato amónico.‒Disolver 2,5 g de vanadato de amonio en 500 ml de agua hirviendo. Después de refrigeración, acidular con 20 ml de ácido nítrico concentrado y diluir hasta 1.000 ml.
7.3.2 Solución de molibdato amónico.‒Disolver 100 g de molibdato amónico en 500 ml de agua a 50 °C. Después de refrigeración añadir con precaución 100 ml de ácido sulfúrico concentrado. Después de enfriar diluir hasta 1.000 ml.
7.3.3 Reactivo molibdato-vanadato.‒Una parte del reactivo 7.3.1 y una parte del reactivo 7.3.2.
7.3.4 Ácido nítrico concentrado.
7.3.5 Ácido sulfúrico concentrado.
7.3.6 Ácido sulfúrico diluido 1:10.
7.3.7 Selenio en polvo.
7.3.8 Agua oxigenada de 20 volúmenes.
7.3.9 Fosfato monopotásico.
7.4 Procedimiento.
7.4.1 Análisis de la muestra:
Introducir en un matraz Kjeldahl 5 g de la sustancia a analizar preparada según el método 1. Añadir 50 ml de H2SO4 concentrado y el catalizador de selenio (utilizando una punta de espátula). Dejar 12 horas en reposo. Añadir unos 20 ml de agua oxigenada. Calentar seguidamente la mezcla hasta ebullición y una vez clarificada prolongarla durante 2 horas de manera que se hidrolice todo el ácido pirofosfórico que hubiera podido formarse, Trasvasar la mezcla después de refrigeración a un vaso volumétrico de 250 ml y completar hasta el trazo de aforo con agua destilada. Si hubiera una precipitación de sulfato de calcio o de ácido salicílico, pasar la solución por un filtro cuantitativo. Para la dosificación del ácido fosfórico, hacer pasar con la pipeta 10 ml de filtrado (el equivalente de 0,2 g) de la muestra en un matraz aforado de 100 ml y añadir 20 ml de ácido sulfúrico diluido y 20 ml de reactivo de molibdato-vanadato. Completar la solución hasta el trazo de aforo con agua y efectuar la medida espectrofotométrica al cabo de 10 minutos.
7.4.2 Preparación de la curva patrón.
Disolver 4,393 g de fosfato monopotásico, previamente secado sobre ácido sulfúrico concentrado, en 1.000 ml de agua. La solución contiene un mg de P por ml. Las soluciones patrón conteniendo de 1 a 45 μg de P por ml se preparan trasvasando con la pipeta la cantidad deseada de solución P en un vaso volumétrico de 100 ml, añadiendo 20 ml de ácido sulfúrico diluido y 20 ml de reactivo de molibdato-vanadato y completando hasta el trazo de calibrado con agua destilada. Al término de 10 minutos se mide la absorción de la solución (en una cubeta de 1 cm de paso) a 436 nm contra una solución testigo, sirviéndose de un espectrofotómetro. Trazar la curva patrón.
7.5 Cálculo.
Calcular el contenido en fósforo total, expresado en porcentaje de P2O5 a partir de la lectura en el espectrofotómetro y con ayuda de la curva patrón.

Porcentaje P2O5 = 2,29 • porcentaje P
Siendo:
A = μg de fósforo leídos en la curva.
M = peso, en g de la muestra.
7.6 Bibliografía.
Pulls, G. (1961) Landwirtschaftliche Forschung, 14, 38-39.
8. CLORUROS
8.1 Principio.
Extracción de los cloruros del producto picado con agua caliente y alcohol y posterior determinación por el método Carpentier-Vohlard.
8.2 Material y aparatos.
8.2.1 Papel de filtro.
8.2.2 Embudos.
8.2.3 Matraces aforados de 250 y 200 ml.
8.2.4 Erlenmeyer de 150 y 250 ml.
8.2.5 Pipetas de 5 ml.
8.2.6 Bureta con divisiones de 0.1 ml.
8.2.7 Balanza analítica.
8.2.8 Placa calefactora.
8.2.9 Agitador magnético con calefacción.
8.2.10 Agitador de imán-teflón.
8.2.11 Vasos de precipitado de 500 ml.
8.2.12 Centrífuga provista de tubos de al menos 100 ml de capacidad.
8.3 Reactivos.
8.3.1 Solución titulada de nitrato de plata 0,1 N.
8.3.2 Solución de ácido nítrico concentrado d = 1,63.
8.3.3 Solución acuosa de sulfato férrico amónico (alumbre férrico) al 4 por 100.
8.3.4 Nitrobenceno.
8.3.5 Solución titulada de sulfocianuro potásico o amónico 0,1 N.
8.3.6 Solución de alcohol etílico al 40 por 100.
8.3.7 Reactivo de Carrez.
8.3.7.1 Solución acuosa de ferrocianuro potásico al 15 por 100.
8.3.7.2 Solución acuosa de acetato de cinc al 30 por 100.
8.4 Procedimiento.
8.4.1 Preparación del extracto.
Pesar, con precisión de 1 mg, un peso P (alrededor de 10 g) de la muestra preparada según el método 1, introduciéndola en un Erlenmeyer de 250 ml. Añadir 150 ml de alcohol al 40 por 100. Poner en el agitador y calentar suavemente. Prolongar la agitación durante una hora al menos. Transvasar a un matraz aforado de 250 ml con alcohol de 40 por 100 a través de un embudo y una varilla. Añadir consecutivamente 5 ml de cada uno de los reactivos de Carrez y enrasar. Agitar y dejar 10 minutos en reposo. Centrifugar 5 minutos a 2.000 r. p m. Separar la grasa que sobrenade con ayuda de una espátula. Filtrar en un matraz aforado de 200 ml hasta el enrase. Verter el contenido del matraz en un vaso de 500 ml, lavando con una pequeña porción de agua. Este líquido de lavado se une al inicial en el vaso. Colocar en una placa y evaporar el líquido hasta 100 ml aproximadamente para eliminar el alcohol. Dejar enfriar y llevar el líquido de nuevo al matraz de 200 ml y enrasar con agua.
8.4.2 Introducir en un Erlenmeyer de 250 ml y agitando suavemente entre adición y adición: 10 ml exactamente medidos de solución 0,1 N de nitrato de plata, 1 ml de solución de ácido nítrico concentrado, 1 ml de solución de sulfato férrico amónico al 4 por 100, 10 ml del extracto problema, 50 ml de agua destilada.
Dejar reposar durante 10 minutos en la oscuridad. Añadir 1 ml de nitrobenceno para aglomerar el precipitado y obtener una solución limpia. Valorar el exceso de nitrato de plata con la solución de sulfocianuro 0,1 N, hasta que se produzca el viraje.
8.5 Cálculos.
El tanto por ciento de cloruros presentes en la muestra, expresado en cloruro sódico, viene dado por la fórmula:

Siendo:
P = peso, en g, de la muestra de la que se ha obtenido el extracto.
n = volumen, en ml, de la solución de sulfocianuro gastados en la valoración.
9. GRASA
9.1 Principio.
Extracción de la grasa de la muestra previamente hidrolizada y desecada, por medio de hexano o éter de petróleo. Eliminación del disolvente por evaporación, desecación del residuo y posterior pesada después de enfriar.
9.2 Material y aparatos.
9.2.1 Erlenmeyer de 500 ml.
9.2.2 Vidrios de reloj.
9.2.3 Placa calefactora.
9.2.4 Papel de filtro Albet 242 ∅ o similar.
9.2.5 Embudos.
9.2.6 Extractor Soxhlet.
9.2.7 Estufa eléctrica.
9.2.8 Balanza analítica.
9.2.9 Desecador provisto de un deshidratante eficaz (gel de sílice) con indicador de humedad.
9.3 Reactivos.
9.3.1 n-hexano o éter de petróleo con un intervalo de punto de ebullición 40-60 °C e índice de bromo inferior a 1, o éter etílico anhidro exento de peróxidos.
9.3.2 Piedra pómez.
9.3.3 Ácido clorhídrico 3 N.
9.4 Procedimiento.
Pesar con aproximación de 1 mg, 2,5 g de muestra preparada como en el método 1 e introducirlos en un Erlenmeyer de 500 ml. Añadir 100 ml de ácido clorhídrico 3 N (9.3.4) y unos trozos de piedra pómez. Cubrir la boca del Erlenmeyer con un vidrio de reloj, y someter la mezcla a una ebullición suave en la placa calefactora durante 1 hora. Enfriar y filtrar sobre doble filtro evitando cualquier paso de materia grasa al filtrado. Lavar el residuo con agua fría hasta desaparición de la reacción ácida. Verificar que en el filtrado no existe materia grasa.
Colocar los papeles de filtro conteniendo el residuo sobre un vidrio de reloj y desecarlos durante hora y media en la estufa a 95-98 °C. Una vez seco el conjunto, introducirlo en el cartucho de extracción, extrayendo con el Soxhlet con éter etílico durante 6 horas, regulando la ebullición de forma que se produzcan 15 sifonadas al menos en cada hora. Eliminar el disolvente en el rotavapor y eliminar el resto del disolvente en la estufa durante hora y media a 75 °C. Enfriar el matraz con la grasa en desecador, matraz que previamente fue tarado, y pesar cuando se alcanza la temperatura ambiente.
Repetir el calentamiento y la pesada hasta que la diferencia entre dos consecutivas sea menor de 5 mg.
9.5 Cálculos.
Expresar el resultado, en porcentaje de peso:

Siendo:
P = peso, en g, del matraz.
P' = peso, en g, del matraz con la grasa.
P" = peso, en g, de la muestra.
9.6 Referencias.
1. Norma ISO-1443.
10. HUMEDAD
10.1 Principio.
Formación de una pasta con ayuda de arena y etanol, que es sometida primeramente a un presecado en baño de María y a continuación secada a 102 ± 2 °C hasta obtener un peso constante.
10.2 Material y aparatos.
10.2.1 Balanza analítica.
10.2.2 Cápsulas de acero inoxidable con tapa de 60 mm de diámetro y 25 mm de altura.
10.2.3 Varilla fina de vidrio con punta aplastada, que entre por completo en la cápsula.
10.2.4 Desecador provisto de un deshidratante eficaz (gel de sílice con indicador de humedad).
10.2.5 Baño de agua.
10.2.6 Estufa eléctrica regulada a 102 ± 2 °C.
10.3 Reactivos.
10.3.1 Arena de mar lavada a los ácidos, cuya granulometría esté comprendida entre 0,25 y 1,4 mm.
10.3.2 Etanol del 95 por 100 en volumen como mínimo.
10.4 Procedimiento.
Secar la cápsula conteniendo una cantidad de arena igual a 3-4 veces el peso de muestra y la varilla de vidrio, durante 30 minutos, en la estufa regulada a 102 ± 2 °C.
Sacarla de la estufa e introducirla en el desecador, hasta que alcance la temperatura ambiente, y pesar el conjunto con 0,1 mg de aproximación.
Introducir en dicha cápsula un peso de muestra preparada según el método 1 aproximadamente 5 g y pesar de nuevo con aproximación de 0,1 mg.
Añadir a la cápsula 5 ml de etanol (10.3.2) y remover la mezcla con la varilla de vidrio. Colocar la cápsula al baño de agua regulándolo a una temperatura comprendida entre 60 y 80 °C para evitar las posibles proyecciones, mantener el calentamiento hasta que el alcohol se evapore. Secar la muestra durante cuatro horas en la estufa a 102 ± 2 °C. Retirar la cápsula de la estufa y colocar en el desecador, hasta que alcance la temperatura ambiente. Pesar con aproximación de 0,1 mg.
Repetir las operaciones de secado hasta peso constante.
Efectuar por lo menos dos determinaciones sobre la misma muestra.
10.5 Cálculos.

Siendo:
Mo = masa, en g, de la cápsula, la varilla y la arena.
M1 = masa, en g, de la cápsula, la varilla, la arena y la muestra antes del desecado.
M2 = masa, en g, de la cápsula, la varilla, la arena y la muestra después del desecado.
10.6 Observaciones.
La diferencia entre los resultados de dos determinaciones simultáneas o realizadas inmediatamente una después de la otra, efectuadas por el mismo analista, no debe ser superior a 0,1 g de agua por 100 g de muestra (0,1 por 100).
10.7 Referencias.
1. Norma internacional ISO R-1442.
11. AZÚCARES TOTALES, REDUCTORES Y LACTOSA
(Método de Luff-Schoorl)
11.1 Principio.
Los azúcares se disuelven en etanol diluido o bien en agua y después de la eliminación del alcohol se valoran por el método de Luff-Schoorl antes y después de la inversión.
11.2 Material y aparatos.
11.2.1 Pipetas de 1, 2, 5, 10, 25 ml.
11.2.2 Probetas de 50 ml.
11.2.3 Matraces Erlenmeyer de cuello esmerilado de 300 ml.
11.2.4 Bureta con divisiones de 0,1 ml.
11.2.5 Refrigerantes de reflujo.
11.2.6 Baño de arena.
11.2.7 Baño de agua.
11.3 Reactivos.
11.3.1 Etanol diluido al 40 por 100 (v/v) d 20 °C ≃ 0,948 llevado a punto de viraje con fenolftaleína en una parte alícuota.
11.3.2 Solución de naranja de metilo al 0,1 por 100 (p/v).
11.3.3 Ácido clorhídrico 4 N.
11.3.4 Ácido clorhídrico 0,1 N.
11.3.5 Hidróxido sódico 0,125 N.
11.3.6 Reactivo de Luff-Schoorl.
11.3.6.1 Solución A.-50 g de ácido cítrico en 500 ml de agua.
11.3.6.2 Solución B.‒Disolver 143,8 g de carbonato de sodio anhidro en 300 ml de agua caliente, dejando enfriar.
11.3.6.3 Solución C.‒Disolver 25 g de sulfato de cobre pentahidrato exento de hierro en 100 ml de agua.
Mezclar cuidadosamente la solución A con la B, añadiendo también con suave agitación la solución C, aforando el conjunto a 1.000 ml. Dejar reposar una noche y filtrar. El pH de la solución debe ser alrededor de 9,4.
Comprobar las concentraciones, que deben ser 0,1 N para el cobre y 2 N para el carbonato sódico.
11.3.7 Solución de tiosulfato sódico 0,1 N con factor exactamente calculado.
11.3.8 Solución de almidón.‒Pesar 5 g de almidón soluble, haciendo un engrudo con 30 ml de agua hirviendo; este engrudo se vierte sobre 1 l de agua hirviendo, manteniendo durante 3 minutos la ebullición, dejar enfriar, y añadir 10 mg de ioduro mercúrico como agente conservador.
11.3.9 Ácido sulfúrico 6 N.
11.3.10 Gránulos de piedra pómez.
11.3.11 Solución hidroglicérida de invertasa, conteniendo 2.750 unidades/ml.
11.3.12 Suspensión de levadura (Saccharomyces cerevisiae) 25 g de dicha levadura fresca en 100 ml de agua. Esta solución se conserva como máximo una semana en el refrigerador.
11.3.13 Soluciones de defecación.
11.3.13.1 Carrez I.‒Disolver en 50 ml de agua 24 g de acetato de cinc (CH3COO)2 Zn.2H2O y 3 g de ácido acético glacial. Completar a 100 ml con agua destilada.
11.3.13.2 Carrez II.‒Disolver en 50 ml de agua 10,6 g de ferrocianuro potásico Fe(CN)6K4.3H2O completando a 100 ml con agua destilada.
11.3.14 Isopentanol como antiespumante (opcional).
11.4 Procedimiento.
11.4.1 Preparación del extracto.
Como en 8.4.1.
11.4.2 Valoración de azúcares reductores.
Tomar con una pipeta una cantidad del extracto no superior a 25 ml y que contenga menos de 60 mg de azúcares reductores expresados en glucosa. Si fuera preciso tomar una cantidad menor de 25 ml (caso no frecuente en productos a base de carne) se completa hasta dichos 25 ml con agua destilada, determinándose el contenido de azúcares reductores según el método de Luff-Schoorl posteriormente descrito.
Los resultados se expresan en glucosa por ciento.
11.4.3 Valoración de azúcares totales, después de la inversión.
Se pueden seguir dos métodos:
11.4.3.1 Método químico.‒Tomar por medio de una pipeta 50 ml del extracto en un matraz de 100 ml aforado, y añadir cuatro gotas de naranja de metilo (11.3.2) y después gota a gota ácido clorhídrico 4 N (11.3.3) hasta viraje neto al color rojo. Añadir posteriormente 15 ml de clorhídrico 0,1 N (11.3.4) y llevar al baño de agua hirviente durante 30 minutos, Enfriar rápidamente a 20 °C y añadir 15 ml de NaOH 0,125 N, o la precisa para el viraje del indicador. Aforar a 100 ml con agua destilada.
Tomar una cantidad no superior a 25 ml que contenga menos de 60 mg de azúcares totales, expresados en glucosa, como en el apartado 11.4.2, determinándose el porcentaje de azúcares reductores según el método de Luff-Schoorl, que se expresa en azúcar invertido o bien en sacarosa, multiplicando el valor obtenido por el factor 0,95.
11.4.3.2 Método enzimático.‒Tomar 50 ml del extracto correspondiente en un matraz de 100 ml aforado, añadir 2 ml de solución hidroglicérida de invertasa y dejar reposar una hora a 50 °C en estufa. Enfriar a 20 °C enrasando exactamente a 100 ml.
Tomar una cantidad no superior a 25 ml y proseguir como en los casos anteriores y con las mismas especificaciones.
11.4.4 Valoración de lactosa.
Tomar en un matraz aforado de 100 ml, 50 ml del extracto correspondiente. Añadir 5 ml de suspensión de levadura (11.3.12), homogeneizar y dejar durante 2 horas en un baño a 39 °C. Enfriar a 20 ºC. Añadir 2,5 ml de solución Carrez I (11.3.13.1) agitar, añadir 2,5 ml de solución Carrez II (11.3.13.2) y agitar de nuevo. Completar el volumen a 100 ml, mezclar y filtrar. Tomar 25 ml de filtrado como máximo, y con un contenido en lactosa entre 40 y 80 mg introducirlos en un Erlenmeyer de 300 ml.
Proceder de la misma forma con un ensayo en blanco de 5 ml de levadura.
Determinar el contenido en lactosa según el método de Luff-Schoorl, los resultados se expresan en lactosa anhidra por ciento.
El método se basa en el hecho de que el Saccharomyces cerevisiae hidroliza todos los azúcares exceptuando la lactosa.
11.4.5 Valoración según el método de Luff-Schoorl.
Tomar con la pipeta 25 ml exactos del reactivo de Luff-Schoorl y verterlos en un Erlenmeyer de 300 ml sobre las correspondientes alícuotas de azúcares, añadir unos gramos de piedra pómez y calentar en tela metálica con mechero fuerte de manera que hierva dos minutos aproximadamente, llevar el Erlenmeyer a un baño de arena previamente calentado de forma que se caliente únicamente el fondo del recipiente, adaptándose un refrigerante de reflujo y manteniendo la ebullición durante 10 minutos exactos.
Enfriar rápidamente, y valorar a los 5 minutos, añadiendo 3 g de ioduro potásico disuelto en 2 ml de agua, y lentamente para evitar las proyecciones, 25 ml de ácido sulfúrico 6 N, valorando el iodo puesto en libertad con tiosulfato 0,1 N, hasta obtener un color amarillo débil, añadir el indicador de almidón (11.3.8) y terminar la valoración hasta decoloración.
Efectuar una valoración análoga en blanco, mezclando 25 ml del reactivo de Luff-Schoorl con 25 ml de agua, sin previa ebullición, y con la adición de ioduro potásico y sulfúrico 6 N.
11.5 Cálculos.
Con la ayuda de la tabla 1 determinar la cantidad de azúcar en mg correspondiente a la diferencia entre los volúmenes de tiosulfato en ml consumidos en la valoración en blanco y problema.
Sean a los mg de azúcares correspondientes:

Siendo:
P = peso, en g, de la muestra inicial de la que se obtuvo el extracto.
V = volumen, en ml, de alícuota tomada.
Esta expresión es válida cuando el extracto se haya enrasado a 250 ml.
11.6 Observaciones.
11.6.1 Los resultados obtenidos para azúcares totales realizando los dos tipos de inversión son semejantes. Pero si el análisis no se realiza con frecuencia, es preferible el método químico, por el posible deterioro de la invertasa y su alto precio.
11.6.2 La diferencia entre el porcentaje de azúcares totales y el de azúcares reductores, multiplicado por 0,95 da el contenido en sacarosa de la muestra.
11.6.3 El contenido en azúcares reductores, exceptuando la lactosa se obtiene multiplicando el valor de ésta, obtenido según (11.4.4), por 0,675 y restando este resultado del contenido en azúcares reductores totales.
TABLA 1
Para 25 ml de reactivo Luff-Schoorl
|
Na2S2O3 0,1 N |
Glucosa, fructosa Azúcares invertidos C8H12O6 |
Lactosa C12H22O11 |
Maltosa C12H22O11 |
|||
|---|---|---|---|---|---|---|
| ml | mg | diferencia | mg | diferencia | mg | diferencia |
| 1 | 2,4 | 2,4 | 3,3 | 3,7 | 3,9 | 3,9 |
| 2 | 4,8 | 2,4 | 7,3 | 3,7 | 7,8 | 3,9 |
| 3 | 7.2 | 2,5 | 11,0 | 3,7 | 11,7 | 3,9 |
| 4 | 9,7 | 2,5 | 14,7 | 3,7 | 15,6 | 4,0 |
| 5 | 12,2 | 2,5 | 18,4 | 3,7 | 19,6 | 3,9 |
| 6 | 14,7 | 2,5 | 22,1 | 3,7 | 23,5 | 4,0 |
| 7 | 17,2 | 2,5 | 25,8 | 3,7 | 27,5 | 4,0 |
| 8 | 19,8 | 2,6 | 29,5 | 3,7 | 31,5 | 4,0 |
| 9 | 22,4 | 2,6 | 33,2 | 3,8 | 35,5 | 4,0 |
| 10 | 25,0 | 2,6 | 37,0 | 3,8 | 39,5 | 4,0 |
| 11 | 27,6 | 2,7 | 40,8 | 3,8 | 43,5 | 4,0 |
| 12 | 30,3 | 2,7 | 44,6 | 3,8 | 47,5 | 4,1 |
| 13 | 33,0 | 2,7 | 48,4 | 3,8 | 51,6 | 4,1 |
| 14 | 35,7 | 2,8 | 52,2 | 3,8 | 55,7 | 4,1 |
| 15 | 38,5 | 2,8 | 56,0 | 3,9 | 59,8 | 4,1 |
| 16 | 41,3 | 2,9 | 59,9 | 3,9 | 63,9 | 4,1 |
| 17 | 44,2 | 2,9 | 63,8 | 3,9 | 68,0 | 4,2 |
| 18 | 47 1 | 2,9 | 67,7 | 4,0 | 72,2 | 4,3 |
| 19 | 50,0 | 3,0 | 71,7 | 4,0 | 76,5 | 4,4 |
| 20 | 53,0 | 3,0 | 75,7 | 4,1 | 80,9 | 4,5 |
| 21 | 56,0 | 3,1 | 79,8 | 4,1 | 85,4 | 4,6 |
| 22 | 59,1 | 3,1 | 83,9 | 4,1 | 90,0 | 4,6 |
| 23 | 62,2 | 88,0 | 94,6 | |||
12. HIDROXIPROLINA
12.1 Principio.
Previa hidrólisis en medio ácido de las proteínas y oxidación de la hidroxiprolina. El derivado formado con el p-dimetilaminobenzaldehido se valora colorimétricamente.
12.2 Material y aparatos.
12.2.1 Balanza analítica.
12.2.2 Matraces de fondo plano de 250 ml de capacidad.
12.2.3 Refrigerantes de reflujo.
12.2.4 Baño de arena.
12.2.5 Agitador magnético.
12.2.6 pH-metro.
12.2.7 Matraces aforados de 50, 100, 200 y 1.000 ml de capacidad.
12.2.8 Erlenmeyer de 250 ml de capacidad.
12.2.9 Tubos de ensayo de 16 x 160 mm, aforados a 12 ml.
12.2.10 Baño de María.
12.2.11 Espectrofotómetro o colorímetro capaces para lecturas a 560 nm.
12.2.12 Cubetas de 1 cm de paso de luz específicas para luz visible.
12.3 Reactivos.
12.3.1 Piedra pómez.
12.3.2 Solución acuosa de ácido clorhídrico al 50 por 100 (v/v) (diluir a 1.000 ml, 500 ml de solución concentrada de ácido clorhídrico de densidad 1,19).
12.3.3 Solución concentrada de hidróxido sódico de densidad 1,33 (400 g/l) (p/v).
12.3.4 Solución de hidróxido sódico al 10 por 100 (p/v).
12.3.5 Alcohol isopropilico puro.
12.3.6 Solución acuosa de cloramina T al 10,5 por 100 (p/v).
12.3.7 Solución tampón de pH = 6 (disolver 34 g de acetato sódico anhidro, 36,5 g de citrato trisódico monohidratado, 5,5 g de ácido cítrico en 385 ml de alcohol isopropílico puro y enrasar a 1.000 ml con agua destilada
12.3.8 Solución oxidante, que debe prepararse en el momento de su empleo, un Volumen de la solución 12.3.6 y cuatro volúmenes de la solución 12.3.7.
12.3.9 Solución acuosa de ácido perclórico al 17,5 por 100.
12.3.10 Solución de p-dimetilaminobenzaldehido (p-DMAB) al 5 por 100 en alcohol isopropilico. Esta solución se conserva una semana a temperatura ambiente.
12.3.11 L-hidroxiprolina.
12.4 Procedimiento.
12.4.1 Preparación de la muestra.
Pesar con 1 mg de aproximación P gramos de la muestra convenientemente homogeneizada (aproximadamente 3 g) sobre papel de fumar sin engomar, e introducir la muestra en el matraz de fondo plano (12.2.2). Añadir piedra pómez 50 ml de la solución de ácido clorhídrico al 50 por 100 (12.3.2). Montar los refrigerantes de reflujo y colocarlos en el baño de arena. Calentar de forma que se mantenga una ebullición suave al menos durante siete horas. Refrigerar rápidamente los matraces bajo corriente de agua. Ajustar el contenido de los matraces a un pH comprendido entre 6 y 7, añadiendo y agitando fuertemente 28 ml de la solución concentrada de hidróxido sódico, dejar enfriar al chorro de agua y llevar al pH anteriormente indicado con solución diluida de hidróxido sódico (12.3.4). Transferir el contenido a un matraz aforado de 200 ml y enrasar dejando en reposo durante una hora. Filtrar a través de papel de filtro plegado. Tomar una alícuota del filtrado y diluirla 10 o 20 veces con agua destilada. Es conveniente realizar ambas diluciones, porque permiten obtener dos valores sobre la misma muestra.
12.4.2 Preparación de la curva patrón.
La coloración obtenida sigue la ley de Beer-Lambert cuando la concentración de hidroxiprolina se encuentra comprendida entre 0 y 20 μg/ml.
Teniendo en cuenta los numerosos factores que influyen en la formación del derivado coloreado, es indispensable construir para cada determinación una curva patrón, realizándose el desarrollo del color al mismo tiempo sobre las diluciones problema y los patrones.
Preparar una solución madre conteniendo 400 μg/ml de hidroxiprolina con agua destilada. A partir de esta solución, hacer diluciones que contengan 5, 10 y 20 μg/ml con agua destilada. Tomar una serie de tubos de ensayo aforados a 12 ml y poner en uno de ellos (tubo testigo) 1 ml de agua destilada. En los tres siguientes colocar 1 ml de las soluciones que contienen 5, 10 y 20 μg/ml de hidroxiprolina. En los dos siguientes 1 ml de cada una de las diluciones diluidas del filtrado. Añadir a cada tubo sucesivamente: 2 ml de isopropanol puro (12.3.5) y 1 ml de la solución oxidante recientemente preparada (12.3.8) y agitar. Dejar reposar durante 10 minutos, transcurrido dicho tiempo añadir de nuevo a cada tubo: 3 ml de la solución de ácido perclórico al 17,5 por 100 (12.3.9) y 2 ml de p-DMAB (12.3.10), homogeneizar el contenido de los tubos y llevarlos al baño de María regulado a 60 °C, permaneciendo en él 20 minutos, retirarlos del baño y enfriarlos balo corriente de agua. Ajustar hasta 12 ml con isopropanol y agitar.
Leer la densidad óptica de cada tubo ajustando el cero con el tubo testigo a 560 nm.
Trazar la correspondiente curva patrón, colocando en abcisas las absorbancias y ordenadas las concentraciones en μg/m1 de hidroxiprolina.
12.5 Cálculos.

Siendo:
x = cantidad de hidroxiprolina leída en la curva patrón.
d = dilución del filtrado realizado.
P = peso inicial de la muestra.
Porcentaje colágeno = 8 % de hidroxiprolina.
12.6 Observaciones.
12.6.1 Cuando la muestra a analizar sea de alto contenido en colágeno, el tiempo de ebullición deberá prolongarse al menos durante nueve horas.
13. NITRITOS
13.1 Principio.
Del extracto obtenido, adicionándole ácido sulfanílico y α-nactilamina, se lee la intensidad de la coloración obtenida mediante colorimetría o espectrofotometría.
13.2 Material y aparatos.
13.2.1 Matraces aforados de 100 y 1.000 ml de capacidad.
13.2.2 Probetas de 100 ó 200 ml de capacidad.
13.2.3 Baño de María.
13.2.4 Papel de filtro plegado de 15 cm de diámetro aproximadamente.
13.2.5 Tubos de ensayo de 150 x 25 mm.
13.2.6 Matraces Erlenmeyer de 300 ml de capacidad.
13.2.7 Espectrofotómetro capaz de leer a 520 nm.
13.2.8 Cubetas de 1 cm de paso específicas para luz visible.
13.3 Reactivos.
13.3.1 Solución patrón de nitrito sódico.‒Pesar, con aproximación de 1 mg, 1 g de nitrito sódico, disolver en agua y completar hasta 1.000 ml. Tomar con la pipeta 5 ml de esta solución e introducirla en otro matraz aforado de 1.000 ml. Enrasar.
13.3.2 Reactivo colorimétrico.
13.3.2.1 Solución L‒Disolver calentando al baño de María 6 g de ácido sulfanílico en 200 ml de ácido acético glacial y 400 ml de agua destilada. Añadir 200 ml de una solución de cloruro sódico que contiene 100 g/l de cloruro sódico. Diluir con agua hasta 1.000 ml.
13.3.2.2 Solución II.‒Disolver calentando al baño de María 0,3 g de cloruro de oc-naftilamina en 100 ml de agua destilada. Filtrar si es necesario y añadir 200 ml de acético glacial. Diluir hasta 1.000 ml con agua destilada.
Manipular con precaución esta disolución por su carácter cancerígeno.
Ambas soluciones (13.3.2.1 y 13.3.2.2) deben conservarse en frascos topacio bien cerrados. El reactivo colorimétrico se obtiene mezclando volúmenes iguales de ambas soluciones.
13.4 Procedimiento.
13.4.1 Preparación del extracto.‒Como en 8.4.1.
13.4.2 Valoración de la muestra.‒Del extracto obtenido en 13.4.1, tomar 25 ml y añadir 1 g, aproximadamente, de carbón activo si es necesario decolorar. Filtrar hasta que el filtrado sea transparente. Del filtrado tomar una alícuota de 10 ml y ponerla en un tubo de ensayo 13.2.5. Si se toman menos de 10 ml, completar hasta 10 ml con agua destilada. Añadir 10 ml del reactivo colorimétrico, mezclar y dejar reposar la solución quince minutos a temperatura ambiente al abrigo de la luz.
A partir de los 20 minutos y antes de 4 horas, medir la densidad óptica de la solución en una cubeta de 1 cm de paso de luz a 520 nm de longitud de onda.
Si la solución coloreada problema presenta una coloración superior a la de la solución patrón más concentrada, tomar una alícuota menor de 10 ml.
Efectuar dos determinaciones sobre la misma muestra.
13.4.3 Preparación de la curva patrón.‒Tomar de la solución patrón (13.3.1) alícuotas de 5, 10 y 20 ml y llevar a 100 ml con agua. El contenido de estas soluciones es, respectivamente, de 0,25, 0,50 y 1 p. p. m. de nitrito sódico.
Transferir 10 ml de cada una de estas soluciones a un tubo de ensayo (13.2.5), añadir 10 ml del reactivo colorimétrico y proceder a su valoración colorimétrica o espectrofotométrica a 520 nm, llevando absorbancias frente a concentraciones expresadas en p. p. m. da nitrito sódico.
13.5 Cálculos.
Calcular el contenido en nitritos de la muestra expresado en p. p. m. por medio de la fórmula:

m = peso de muestra de la que se ha obtenido el extracto.
V = volumen, en ml tomado del extracto decolorado.
C = concentración en nitrito sódico expresada en μg/ml determinada sobre la curva patrón.
Tomar como resultado la media de los valores obtenidos en dos determinaciones paralelas si las condiciones de reproducibilidad se cumplen.
13.6 Observaciones.
La diferencia entre dos determinaciones paralelas sobre una misma muestra, realizadas simultáneamente por el mismo analista, no debe ser superior al 10 por 100 del contenido calculado en nitritos.
13.7 Referencias.
1. Norma ISO/DIS 2918.
14. NITRATOS
14.1 Principio.
Al reaccionar en medio sulfúrico los nitratos con la brucina se produce una coloración amarilla-marrón, cuya intensidad es proporcional al contenido en nitratos presentes, lo que permite su valoración colorimétrica o espectrofotométrica.
14.2 Material y aparatos.
14.2.1 Matraz aforado de 50 ml de capacidad.
14.2.2 Espectrofotómetro o colorímetro capaces para lecturas a 410 nm.
14.2.3 Cubetas de 1 cm de paso específicas para luz visible.
14.3 Reactivos.
14.3.1 Solución patrón de nitratos.‒Disolver 0,1629 g de nitrato potásico anhidro en agua y enrasar a 1 l. 1 ml de esta solución contiene 0,1 mg de nitrato.
14.3.2 Reactivo brucina-Ácido sulfanílico.‒Disolver 1 g de brucina y 0,1 g de ácido sulfanílico en unos 70 ml de agua destilada caliente, añadir 3 ml de ácido clorhídrico concentrado, dejar enfriar y enrasar a 100 ml con agua. La solución debe guardarse en frasco color topacio.
14.3.3 Solución de ácido sulfúrico.‒Añadir con cuidado 500 ml de ácido sulfúrico concentrado a 75 ml de agua destilada. Debe conservarse herméticamente cerrado.
14.4 Procedimiento.
14.4.1 Preparación del extracto.‒Como en 8.4.1.
14.4.2 Valoración de la muestra.‒Introducir en un matraz aforado de 50 ml de capacidad 10 ml del extracto (14.4.1), 1 ml del reactivo (14.3.2) y 10 ml del reactivo (14.3.3) muy lentamente, mezclar y dejar en reposo durante diez minutos al abrigo de la luz.
Pasado este tiempo, añadir agua destilada, agitando, hasta completar unos 40 ml y dejar reposar durante quince minutos en la oscuridad.
Enfriar el matraz en un baño de hielo hasta que alcance la temperatura ambiente, preferiblemente también en la oscuridad. A continuación, enrasar a 50 ml con agua, homogeneizar el contenido del matraz y leer la absorbancia a 410 nm. El cero se ajusta con 20 ml de agua sometida al proceso anteriormente descrito.
14.4.3 Preparación de la curva patrón. Tomar distintas alícuotas de la solución 14.3.1 y tratar como en 14.4.2.
14.5 Cálculos.
Llevar la absorbancia obtenida a la curva patrón y expresar el correspondiente contenido de nitratos en mg por kg.
14.6 Observaciones.
14.6.1 Periódicamente resulta aconsejable confirmar o rectificar los valores de la curva patrón, lo que es absolutamente necesario cuando se cambia de reactivos.
14.6.2 En las condiciones citadas, se pueden medir concentraciones de nitrato comprendidas entre 50 y 500 μ g/l.
14.6.3 El ácido sulfanílico se añade al reactivo 14.3.2 para evitar la posible interferencia del ion nitrito. Las interferencias de iones ferrosos, férricos y manganosos sólo son apreciables cuando su concentración es superior a 1 mg/l.
15. pH
15.1 Principio.
Medida del potencial eléctrico creado en la membrana de un electrodo de vidrio, función de la actividad de iones hidrógeno a ambos lados de la membrana. Utilizar como referencia un electrodo de calomelanos.
15.2 Material y aparatos.
15.2.1 pH-metro capaz de determinar la segunda cifra decimal.
15.2.2 Electrodos de vidrio de punta fina o convencional (según la dureza de la muestra).
15.2.3 Electrodo de calomelanos.
15.3 Reactivos.
15.3.1 Disolución tampón de pH = 4.
15.3.2 Disolución tampón de pH = 7
15.4 Procedimiento.
Tomar una cantidad de muestra preparada según el método 1 o bien directamente, en el caso de muestras homogéneas, añadir igual cantidad de agua destilada, mezclar y dejar reposar durante diez minutos.
Ajustar el pH-metro a la temperatura de trabajo con las disoluciones 15.3.1 y 15.3.2 e introducir los electrodos de vidrio y de referencia, separados a. menos 2 cm.
Conectar el pH-metro y esperar a que se estabilice.
15.5 Expresión de los resultados.
Anotar el valor de pH indicado por el aparato.
Repetir la operación dos veces al menos.
15.6 Observaciones.
15.6.1 Es necesaria una limpieza escrupulosa de los electrodos entre determinaciones sucesivas, lo que al tratarse de productos con un alto contenido en materia grasa es más laboriosa que en disoluciones desengrasadas, requiriendo a veces ser frotados o sumergidos en alcohol, éter dietílico, éter de petróleo, etcétera; secarlos convenientemente y lavarlos finalmente con agua destilada; secarlos de nuevo, quedando así dispuestos para una nueva lectura.
MÉTODOS BIOLÓGICOS
1. IDENTIFICACIÓN DE ESPECIE ANIMAL
1.1 Principio.
Las proteínas musculares de las distintas especies animales, aunque muy similares entre sí, tienen en cada grupo zoológico una estructura específica que permite diferenciar carnes de especies distintas por métodos serológicos (técnica de Uhlenhuth).
Sólo es aplicable a productos crudos.
1.2 Material y aparatos.
1.2.1 Tubos de precipitación de 20 x 4 mm.
1.2.2 Gradillas metálicas para los tubos anteriores.
1.2.3 Precipitoscopio (cámara oscura con luz indirecta).
1.2.4 Pipetas capilares muy finas.
1.2.5 Pinzas.
1.2.6 Bisturí.
1.2.7 Tijeras.
1.2.8 Gasa.
1.2.9 Papel de filtro.
1.2.10 Tubos de ensayo.
1.2.11 Erlenmeyer de 250 ml de capacidad.
1.3 Reactivos.
1.3.1 Suero fisiológico estéril.
1.3.2 Antisueros específicos (su especificidad debe ser contrastada en el momento de su utilización).
1.4 Procedimiento.
Separar del producto cárnico entre 5 y 10 g de carne magra lo más exenta posible de grasa. Picar en trozos muy pequeños, poniéndose a macerar en suero fisiológico estéril en la proporción de 1/10 Mantener el macerado puesto en un Erlenmeyer de 250 ml en frigorífico durante 24 horas, al objeto de evitar la proliferación bacteriana.
Transcurridas las 24 horas, filtrar el macerado a través de gasa y papel de filtro cuantas veces sea necesario, hasta obtener un líquido transparente, sin importar que tenga coloración propia. Diluir el filtrado, que estaba a 1/10, hasta 1/25 y 1/50.
Poner en una gradilla metálica tres tubos de precipitación para las tres diluciones preparadas. Tomar con pipeta capilar una para cada dilución, partes de las disoluciones (antígeno problema), llevándolo hasta el fondo de los tubos. Colocar seguidamente el antisuero que se ensaya (anticuerpo específico conocido) en los tres tubos mediante pipeta capilar muy fina de la manera siguiente: Poner el tubo conteniendo el antígeno problema en posición horizontal, llevando en este momento la pipeta con el antisuero hasta el fondo del tubo, momento en que se vuelve a poner el tubo en posición vertical, con le que saldrá una pequeña cantidad de antisuero que desplazará hacia arriba el antígeno, pero sin mezclarse con él; volver a poner de nuevo en posición horizontal el tubo, sacando rápidamente la pipeta que contiene el antisuero. El tubo debe estar siempre en posición horizontal a la entrada o la salida de la pipeta con el antisuero, evitando de esta forma que los dos líquidos reaccionantes se mezclen. Utilizar una sola pipeta para cada antisuero. Es conveniente poner dos tubos testigos, uno para el suero y otro para el antígeno, testigos que necesariamente deben dar siempre reacción negativa.
1.5 Interpretación de resultados.
La reacción positiva se produce con la aparición de un anillo blanquecino, opalescente, en la zona de separación de ambos líquidos reaccionantes antes de 15 minutos. Reacciones positivas posteriores a este tiempo deben considerarse inespecíficas. La reacción tiene lugar, para un mismo antígeno, a distintos tiempos, según sea el título del antisuero empleado.
1.6 Observaciones.
1.6.1 La lectura de los resultados debe hacerse con luz intensa indirecta y sobre fondo oscuro, preferiblemente en un precipitoscopio.
1.7 Referencias.
1. Normalización de Técnicas en Bromatología Sanitaria. F. Pérez Flórez. 1970.
Métodos de análisis de cereales y derivados
16. ÁCIDO ASCÓRBICO (VITAMINA C)
(Método cualitativo)
16.1 Principio.
Este método sirve para determinar la presencia de vitamina C en harina y consiste en la aparición de puntos blancos sobre fondo rosa del reactivo sal sódica del 2,6 diclorofenol indofenol en medio ácido.
16.2 Material y aparatos.
16.2.1 Placa Petri de 65 cm2.
16.3 Reactivos.
16.3.1 Disolución acuosa al 0,05 por 100 de la sal sódica del 2,6 diclorofenol indofenol.
16.3.2 Disolución acuosa al 5 por 100 de ácido metafosfórico.
16.4 Procedimiento.
Extender 10 g de la muestra sobre una placa de vidrio compactándola de forma que quede bien uniforme. Rociar por completo con la disolución del ácido metafosfórico y a continuación hacer lo mismo con la disolución de la sal sódica del 2,6 diclorofenol indofenol. Al cabo de unos minutos aparecen unos puntos blancos más o menos grandes sobre el fondo rosa.
18. FÓSFORO
18.1 Principio.
Transformación de los compuestos fosforados en ortofosfatos y posterior valoración colorimétrica con fosfomolibdovanadato.
18.2 Material y aparatos.
18.2.1 Espectrofotómetro o colorímetro que permita lecturas a 430 nm.
18.2.2 Crisoles de porcelana, de 35 mm de diámetro y 45 milímetros de altura, sin tapadera.
18.2.3 Matraces aforados de 100 y 500 ml de capacidad.
18.2.4 Baño de agua.
18.2.5 Estufa de desecación con sensibilidad de ± 1 ºC.
18.3 Reactivos.
18.3.1 Amoníaco concentrado de densidad ρ = 0,910 g/ml.
18.3.2 Disolución de molibdato amónico al 10 por 100 (p/v). Disolver 100 g de molibdato amónico en agua caliente; añadir 10 ml de amoníaco concentrado de ρ = 0,910 g/cm3 para asegurar su conservación y completar hasta 1.000 ml con agua destilada.
18.3.3 Ácido nítrico de densidad ρ = 1,38 g/ml.
18.3.4 Ácido nítrico al 10 por 100 (p/v).‒Disolver 16 ml de ácido nítrico ρ = 1,38 g/cm3, hasta 100 ml con agua destilada.
18.3.5 Disolución de metavanadato de amonio.‒Disolver 2,35 gramos de metavanadato de amonio en 400 ml de agua destilada y caliente. Añadir lentamente y agitando 20 ml de la disolución que contiene 7 ml de ácido nítrico ρ = 1,381 g/ml y 13 ml de agua destilada.
18.3.6 Reactivo nitromolibdovanadato.‒Mezclar 200 ml de la disolución de molibdato amónico al 10 por 100 y 200 ml de la disolución de metavanadato de amonio, con 134 ml de ácido nítrico ρ = 1,381 g/cm3 o en su lugar 192 ml de ácido nítrico ρ = 1,331 g/ml.
18.3.7 Ácido clorhídrico concentrado de densidad ρ = 1,19 g/ml.
18.3.8 Disolución patrón de fósforo.‒Pesar 4,394 g de fosfato monopotásico, previamente desecado en estufa a 100 °C durante unas doce horas. Disolver en agua destilada y llevar a un volumen de 1.000 ml en un matraz aforado, 1 ml corresponde a 1.000 gammas de fósforo
18.3.9 Disoluciones patrones.‒Tomar 10 ml de la disolución anterior y diluir con agua destilada hasta el enrase en un matraz aforado de 100 ml, obteniéndose una concentración de 100 gammas de fósforo por mililitro. Tomar partes alícuotas de 0,5, 1,0, 1,5, 2,0 y 2,5 ml y llevar a un volumen de 10 ml con agua destilada. Su concentración será de 5, 10, 15, 20 y 25 gammas /ml. Añadir 10 ml del reactivo nitromolibdovanadato y proceder como se describe en el método.
18.4 Procedimiento.
Pesar, con una aproximación de ± 0,1 mg, de 0,3 a 1,5 g de muestra en un crisol previamente calcinado y tarado. Introducir el crisol en la mufla a una temperatura inferior a 100 °C. Aumentar la temperatura paulatinamente hasta alcanzar los 550 °C; mantener esta temperatura durante 2 horas. No se debe pasar de 550 °C, para evitar decrepitaciones y volatilizaciones, ya que cuando existen cloruros en el producto a analizar, puedan afectar a los resultados. El tiempo que debe permanecer el crisol con la muestra en la mufla es variable y estará de acuerdo con la naturaleza y con la cantidad de la muestra. Suelen ser suficientes 2 horas, pero si transcurrido este tiempo las cenizas del crisol no presentan el color blanco grisáceo deseado, sacar el crisol de la muña y dejar enfriar dentro de un desecador, añadiendo posteriormente unas gotas de agua o, mejor, unas gotas de agua oxigenada de 50 volúmenes, introducir el crisol en la estufa de desecación a 100 °C para eliminar el agua o el agua oxigenada. Eliminada la humedad, introducir nuevamente el crisol en la mufla a 550 °C. Dejar transcurrir el tiempo necesario hasta que las cenizas contenidas en el crisol alcancen el color deseado. Sacar el crisol de la mufla y llevar a un desecador con sustancias desecadoras. Dejar enfriar hasta la temperatura ambiente.
Obtenidas las cenizas, añadir en el crisol una cantidad de ácido clorhídrico concentrado (18.3.7) hasta que las cenizas queden cubiertas Evaporar el ácido clorhídrico en el baño de agua a ebullición, hasta sequedad, en un dispositivo adecuado para la eliminación de vapores ácidos. Disolver el residuo en 3 ml de ácido nítrico al 10 por 100 y hervir en el baño de agua durante 5 minutos, utilizando el dispositivo adecuado para la eliminación de los vapores ácidos. No se debe dejar secar el contenido para evitar la hidrólisis de los ortofosfatos que produciría reacciones coloreadas Filtrar a través de papel, sobre un matraz aforado de 500 ml, lavando con agua destilada los crisoles donde estaban contenidas las cenizas y diluir hasta el enrase.
Para desarrollar la reacción de color, colocar, en una cubeta o tubo de espectrofotómetro o fotocolorímetro, 10 ml de la disolución problema y añadir 10 ml del reactivo nitromolibdovanadato. Agitar, dejar reposar durante diez minutos. Efectuar la lectura espectrofotométrica o fotocolorimétrica a 430 nm, utilizando como blanco la mezcla de 10 ml de agua destilada y 10 ml de reactivo nitromolibdovanadato. La coloración amarilla desarrollada es estable durante varios días.
18.5 Cálculos.
Leer en el espectrofotómetro o fotocolorímetro y buscar su correspondencia en fósforo en la curva patrón.

Siendo:
F = concentración de fósforo, en gammas, encontrada en la curva patrón/10 ml disolución.
P = peso, en gramos, de la muestra empleada.
18.6 Referencias.
1. Instituto de Racionalización y Normalización del Trabajo. Una Norma Española UNE 64017.
19. DETECCIÓN Y CUANTIFICACIÓN DE HARINAS DE TRIGOCOMÚN (TRITICUM VULGARE) EN SÉMOLAS Y PASTAS ALIMENTICIAS
19.1 Principio.
El método se basa en la detección y cuantificación de un componente designado CM2, del extracto cloroformo-metanol del endospermo de trigo o de productos derivados de él, cuyo control genético radica en el cromosoma 1D de «Triticum Vulgare» y que por tanto no se encuentra en «T. durum».
El mencionado extracto, al que se designa proteína CM, se fracciona por electroforesis sobre gel de almidón, y el componente CM1 se estima visualmente o se cuantifica densitométricamente.
19.2 Material y aparatos.
19.2.1 Balanza analítica.
19.2.2 Equipo de electroforesis.‒Fuente de tensión, corriente continua 0-500 V, ‒100 mA. Placas de vidrio de 20 x 20 cm marcos de plástico de 20 x 20 cm exterior, de 18 x 18 cm interior y de 3 mm de espesor. Depósitos de electrodos de 20 x 10 x 10 centímetros.
19.2.3 Densitómetro de reflexión en el caso de que se quiera cuantificar.
19.2.4 Tubos de 8 x 50 mm o similar, con tapones de corcho.
19.2.5 Gradilla.
19.2.6 Dos placas de acero inoxidable de 5 x 7 x 1 cm o dimensiones similares.
19.2.7 Jeringa de vidrio de 1 ml de capacidad.
19.2.8 Capilares de 10 cm de largo.
19.2.9 Placa de porcelana con pocillos.
19.2.10 Probetas de 100 ml y 1.000 ml.
19.2.11 Vasos de 500 ml y de 1.000 ml.
19.2.12 Baño de agua con termómetro.
19.2.13 Cubeta de plástico de al menos 25 x 25 x 5 cm.
19.3 Reactivos.
19.3.1 Cloroformo.
19.3.2 Metanol.
19.3.3 Éter dietílico libre de peróxidos.
19.3.4 Etanol del 70 por 100.
19.3.5 Papel Albet número 502, o similar, y papel de filtro.
19.3.6 Almidón hidrolizado para electroforesis Connauglit o similar.
19.3.7 Tampón lactato de aluminio-ácido láctico, 0,1 N, pH 3,2 en urea 3 M.‒Diluir 4,9 g de lactato de aluminio en agua destilada, añadir 10,6 ml de ácido láctico purísimo y 180 g de urea, completando hasta 1 litro con agua destilada. Este tampón sirve tanto para el gel de almidón como para los compartimentos de los electrodos.
19.3.8 Solución de nigrosina soluble en agua al 0,5 por 100 en acético-agua (1/1) (v/v).
19.3.9 Gel de almidón.‒Mezclar 24,5 g de almidón y 180 ml de tampón en un vaso de 500 ml, agitar suavemente con una varilla en baño de agua a 80 ± 3 ºC hasta que gelifique (2-3 minutos). El gel caliente se vierte sobre una placa de vidrio a la que se ha superpuesto un marco de plástico de las mismas dimensiones externas que la placa y de 3 mm de espesor, extender uniformemente con la varilla y, finalmente, prensar suavemente con una placa de vidrio de las mismas dimensiones sin dejar burbujas. Dejar reposar durante al menos 3 horas.
19.4 Procedimiento.
19.4.1 Extracción y preparación para electroforesis de la proteína CM.‒Pesar 50 mg de harina, sémola, grano o pasta alimenticia y transferirlos o un tubo de 8 x 50 mm o similar. El grano y la pasta se aplastan por presión entre dos placas de acero inoxidable antes de ser transferidas al tubo. Añadir aproximadamente 0,5 ml de éter dietílico en cada tubo y dejar reposar durante no menos de 30 minutos, agitando ocasionalmente. Después de la última agitación se deja sedimentar por gravedad y el sobrenadante se elimina con la ayuda de una jeringa. El disolvente residual se deja evaporar a la temperatura ambiente o en una estufa a 35 °C durante 10 minutos. Agregar aproximadamente 0,25 ml de cloroformo-metanol (2/1) (v/v) a cada tubo, tapar y dejar reposar durante 2 horas, agitando ocasionalmente.
Después de la última agitación, dejar sedimentar por gravedad y transferir el extracto sobrenadante con un capilar a una pieza de papel Albet número 502 o similar, de dimensiones 3 x 10 mm. Con el capilar se satura el papel, que se deja evaporar antes de una nueva adición (ver 19.6.4), repitiendo la operación hasta agotar el sobrenadante. Para esta operación, las piezas de papel a las que se van a transferir las distintas muestras se depositan en distintos pocillos de una placa de porcelana o similar Se recomienda realizar la transferencia entre 10 y 20 muestras simultáneamente.
19.4.2 Electroforesis sobre gel de almidón de la proteína CM.‒Separar cuidadosamente una de las placas de vidrio con la ayuda de una espátula y recubrir la superficie expuesta del gel con un plástico fino. Marcar en dicha superficie una fila de ranuras (1 cm de largo cada una) a 3 cm de uno de los bordes del gel. Alojar en dicha ranura las piezas de papel Albet número 502 o similar que portan las muestras, previamente impregnadas con tampón. Disponer el gel horizontalmente apoyado sobre las cubetas de electrodos y establecer la conexión eléctrica mediante puentes de papel de filtro (20 papeles de dimensiones apropiadas superpuestos).
19.5 Interpretación de resultados.
19.5.1 Detección de trigo exaploide.‒La electroforesis del extracto CM de trigo exaploide («T. Vulgare») muestra tres bandas designadas CM1, CM2 y CM3, mientras que la de trigo tetraploide («T. durum») sólo presenta dos CM2 y CM3.
El trigo exaploide se detecta en una mezcla por la aparición de CM1 en el perfil electroforético. Para el tamaño de muestra anteriormente propuesto, CM1 se detecta en mezcla de 10-15 por 100. Usando muestras de tamaño doble, el umbral de detección se reduce proporcionalmente.
19.5.2 Cuantificación de trigo exaploide en mezclas.‒La acotación del porcentaje de «T. Vulgare» en una mezcla se basa en la cuantificación de la relación CM1, CM2 y en la estimación de la variabilidad intraespecífica de CM1 y CM2.
En la figura 1-A se presenta la forma de acotar gráficamente el porcentaje de trigo exaploide en mezclas basándose en la medida de la relación CM1, CM2 mediante densitometría de reflectancia con luz de 620 mm. En la figura 1-B se representa la variación de la amplitud de la acotación según el valor obtenido.
Una estimación semicuantitativa, más imprecisa que la anterior, puede obtenerse por comparación visual del problema con una serie de mezclas conocidas que pueden incorporarse al mismo gel.
19.6 Observaciones.
19.6.1 Las condiciones de electroforesis son 10 V/cm durante seis horas.
19.6.2 La tinción se realiza con nigrosina al 0,05 por 100 en acético.‒Agua (1/1) (v/v) durante 14-16 horas en una cubeta de plástico de dimensiones apropiadas, dejando el gel con la cara opuesta a la de inserción hacia arriba.
19.6.3 La decoloración del fondo se realiza en pocos minutos con etanol al 70 por ICO.
19.6.4 Cuando se manejan 10-20 muestras, después de una transferencia se devuelve el capilar al tubo y se pasa a la muestra siguiente. Cuando se llega a la muestra final, ya se ha evaporado la primera y está en condiciones de una nueva transferencia.
19.7 Referencias.
1. R. García Faure y F. García Olmedo: «A new Method for the Estimation of Common Wheat in Pasta Products». Lebensm. Wis. U. Technol. Vol. 2. 1969.
20. DETECCIÓN DE HARINAS DEGRADADAS POR EL ATAQUE DE PENTATÓMIDOS
20.1 Principio.
Se detecta la degradación de la calidad panadera de la masa de harina mediante la determinación del exceso de actividad proteolítica.
20.2 Material y aparatos.
Como en 14.2.
20.3 Reactivos.
Como en 14.3.
20.4 Procedimiento.
Como el 14.4 con las siguientes modificaciones:
20.4.1 El número de piezas de masa serán seis.
20.4.2 Transcurrido el tiempo normal de 26 minutos del comienzo de amasado, extraer tres piezas de la cámara del alveógrafo y analizarlas obteniendo sus correspondientes curvas. El resto de las piezas se analizan sobre el mismo papel después de un periodo de reposo de tres horas.
Si alguno de los alveolos o curvas fuera claramente anormal debe desecharse la curva.
20.5 Expresión de los resultados.
Si existe una actividad proteolítica excesiva, la segunda serie de curvas presentará menor extensibilidad y tenacidad, siendo mayor la diferencia entre ellas, a mayor actividad.
Cuantificar esta actividad calculando la degradación de W y G en la forma siguiente:
Calcular los valores de estos índices por separado para la primera serie de curvas (con tiempo de reposo normal) Wo y Go y para la segunda (con tiempo de reposo de 3 horas) W1 y G1.


20.6 Referencias.
1. Harinas. Actividad proteolítica. H-80277-A. Ministerio del Aire.
16(b). FÓSFORO SOLUBLE EN AGUA
(Colorimetría)
16(b).1. Principio.
Extraer el fósforo soluble en agua de la muestra y determinar su cantidad midiendo espectrofotométricamente la absorbancia del complejo coloreado que forma el ion ortofosfato con el molibdeno.
No es aplicable a las escorias básicas ni a los abonos que producen soluciones coloreadas o que contengan otros iones además del ortofosfato que formen complejos coloreados con el malibdovanadato.
16(b).2. Material y aparatos.
16(b).2.1 Matraces aforados de 100 ml de capacidad.
16(b).2.2 Buretas de 20 ml de capacidad.
16(b).2.3 Pipetas de 20 ml de capacidad.
16(b).2.4 Espectrofotómetro o colorímetro capaces para lecturas a 400 nm.
16(b).3. Reactivos.
16(b).3.1 Soluciones de fosfato monopotásico puro y seco (a 105 °C durante dos horas) que contengan de 0,4 a 1,0 mg de P2O5/ml con intervalos de 0,1 mg/ml. Preparar semanalmente las soluciones de 0,4 a 0,7 mg de P2O5/ml.
16(b).3.2 Ácido nítrico.
16(b).3.3 Solución de molibdovanadato.‒Disolver 40 g de molibdato vanadato amónico tetrahidrato en 400 ml de agua caliente y enfriar. Disolver 2 g de metavanadato amónico en 250 ml de agua caliente, enfriar y añadir 450 ml de ácido perclórico del 70 por 100. Añadir gradualmente y agitando la solución de molibdato a la solución de metavanadato y diluir a 2 1.
16(b).4. Procedimiento
16(b).4.1 Preparación de la solución.‒Poner un gramo de muestra sobre un filtro de 9 cm de diámetro y lavar con pequeñas porciones de agua hasta obtener, aproximadamente, 250 ml de filtrado. Dejar pasar cada porción a través del filtro antes de añadir otra nueva y si el filtrado no puede completarse al cabo de una hora, aplicar succión. Si el filtrado está turbio añadir de 1 a 2 ml de ácido nítrico. Enrasar a 250 ml con agua destilada y mezclar. Para muestras con menos del 5 por 100 de P2O5 diluir a 250 ml y para muestras que contengan más del 5 por 100 de P2O5 diluir a un volumen tal que una alícuota de 5 ó 10 ml contenga de 2 a 5 mg de P2O5.
16(b).4.2 Muestras que contienen menos del 5 por 100 de P2O5.‒Introducir en un matraz aforado de 100 ml una alícuota de 5 ml de la solución problema [16(b).4.1] y 5 ml de la solución patrón de fosfato que contenga 2 mg de P2O5. Desarrollar color como en 16(b).5.1 y determinar la absorbancia correspondiente de la misma manera que para las soluciones patrones de fosfatos 16(b).6.1, ajustando el aparato a cero de absorbancia con el patrón de 2 mg. Leer el contenido de P2O5 de la solución problema llevando el valor de la absorbancia a la curva patrón [16 (b).6.1].
Como series de soluciones problemas, vaciar y rellenar la célula de referencia con el patrón de 2 mg después de cada determinación.
16(b).4.3 Muestras que contienen más de 5 por 100 de P2O5.‒Tomar alícuota de 5 ó 10 ml de la solución problema (para que contenga de 2 a 5 mg de P2O5) [16(b) 4.1] e introducir en un matraz aforado de 100 ml sin adición de solución patrón de fosfato y proceder como en 16(b).4.2.
16 (b).5. Cálculo.
16 (b).5.1 Preparación de la curva patrón.‒Tomar alícuotas de los siete patrones de fosfato monopotásico [16(b).31] (conteniendo de 2-5 mg de P2O5/alícuota) en matraces aforados de 100 ml y añadir 45 ml de agua. A continuación, y dentro de cinco minutos cuando se trata de series, añadir 20 ml de molibdovanadato con una bureta o pipeta, enrasar y mezclar. Dejar reposar diez minutos. Leer a 400 nm.
16 (b).5.2 Muestras que contienen menos de 5 por 100 de P2O5:

16 (b).5.3 Muestras que contienen más de 5 por 100 de P2O5:

16 (b).6. Observaciones.
16(b).6.1 Introducir en matraces aforados de 100 ml alícuotas de 5 ml de las soluciones de fosfato 16 (b).3.1 que contenga 2 y 3,5 mg de P2O5/alícuota, respectivamente, y desarrollar color como en 16(b).5.1. Ajustar el aparato poniendo a cero la absorbancia con la solución patrón de 2 mg y determinar la absorbancia para la solución patrón de 3,5 mg (esencial que la absorbancia de esta última sea prácticamente idéntica al valor que le corresponde en la curva patrón).
16(b).7. Referencias.
1. A. O. A. C.: «Official Methods of Analysis». Ed. 1970, página 14/20.032.
17(b). FÓSFORO SOLUBLE EN AGUA Y EN CITRATOAMÓNICO NEUTRO
(Asimilable) (Colorimetría)
17(b).1. Principio.
Después de separar el fósforo soluble en agua de la muestra, someter el residuo a una extracción con disolución neutra de citrato amónico. El fósforo asimilable puede determinarse en la disolución obtenida al reunir los extractos acuosos y de citrato amónico.
No es aplicable a aquellos abonos que después de tratarlos con la mezcla ácida ternaria conserven algo de color o aquellos que contengan otros iones además del ortofosfato que formen complejos coloreados con el molibdovanadato.
17(b).2. Material y aparatos.
17(b).2.1 Matraces aforados de 500 ml de capacidad.
17(b).2.2 Matraces aforados de 200 ml.
17(b).2.3 Matraces aforados de 250 ml.
17(b).2.4 Espectrofotómetro o colorímetro capaces para lecturas a 400 nm.
17(b).2.5 Placa calefactora.
17(b).3 Reactivos.
17(b).3.1 Soluciones de fosfato monopotásico puro y seco (a 105 °C durante dos horas) que contengan de 0,4 a 1,0 mg de P2O5/ml con intervalos de 0,1 mg/ml. Preparar semanalmente las soluciones de 0,4 a 0,7 mg de P2O5/ml.
17(b).3.2 Mezcla ácida ternaria.‒Añadir 20 ml de ácido sulfúrico a 100 ml de ácido nítrico, mezclar y añadir 40 ml de ácido perclórico al 70 por 100.
17(b).3.3 Molibdovanadato modificado.‒Disolver 40 g de molibdato amónico en 400 ml de agua caliente y enfriar. Disolver 2 g de metavanadato amónico en 250 ml de agua caliente, enfriar y añadir 250 ml de ácido perclórico del 70 por 100. Añadir gradualmente agitando la solución de molibdato a la solución de metavanadato y enrasar a 2 litros.
17(b).3.4 Solución de citrato amónico neutro.‒Debe tener un peso específico de 1,09 a 20 °C y un pH igual a 7,0 determinado electrométricamente.
Disolver 370 g de ácido cítrico cristalizado en 1,5 litros de agua y casi neutralizar, añadiendo 345 ml de hidróxido amónico (de 28 a 29 por 100 de NH3). Si la concentración de amoníaco es menor de) 28 por 100 añadir mayor volumen y disolver el ácido cítrico en un volumen más pequeño de agua. Enfriar y comprobar el pH. Ajustar a pH = 7 con hidróxido amónico (1 + 7) o con disolución de ácido cítrico. Si es preciso diluir la disolución hasta que el peso específico sea de 1,09 a 20 °C. El volumen será, aproximadamente de 2 litros.
Conservar en frascos herméticamente cerrados y comprobar el pH de vez en cuando, reajustándolo si difiere de 7,0.
17(b).4. Procedimiento.
17(b).4.1 Preparación de la solución.‒Poner un gramo de muestra sobre un papel de filtro de 9 cm de diámetro y lavar por gravedad sobre un matraz aforado de 500 ml con 12 porciones de agua de 10 ml. Dejar que cada porción pase a través del filtro antes de añadir más. Dejar escurrir el papel completamente y después enjuagar el embudo con lo ml de agua destilada.
Tratar el residuo insoluble en agua con la solución de citrato amónico neutro, transfiriendo el filtro y el residuo, dentro del intervalo de una hora, a un matraz de 200 ó 250 ml que contenga 100 ml de citrato amónico previamente calentado a 65 °C Cerrar herméticamente el matraz con un tapón de goma blanda, agitar vigorosamente hasta reducir a pulpa el papel y quitar la presión levantando momentáneamente el tapón. A continuación colocar el matraz cerrado en un baño agitador a 65 °C exactamente y agitar durante una hora (la acción del aparato debe ser tal que la dispersión de la muestra ha de ser mantenida continuamente en la solución de citrato y que esta solución bañe continuamente las superficies internas del tapón y del matraz). Quitar el matraz del aparato y transferir su contenido al que tiene la fracción soluble en agua. Enfriar a temperatura ambiente inmediatamente enrasar y mezclar completamente y dejar en reposo por lo menos dos horas antes de tomar la alícuota.
17(b).4.2 Muestras que contienen menos del 5 por 100 de P2O5.‒Tomar una alícuota de 10 ml en un matraz Erlenmeyer de 125 ml. Añadir 5 ml de mezcla ácida ternaria, agitar el matraz con movimiento rotatorio, hervir suavemente durante quince minutos y digerir a 150-200 °C hasta que la solución quede con una sal blanca e incolora. Evaporar hasta humos blancos y continuar calentando durante cinco minutos. Enfriar y añadir 15 ml de agua y hervir cinco minutos. Trasvasar el contenido del Erlenmeyer a un matraz aforado de 100 ml, diluir a 50 ml, agitar con movimientos rotatorios y enfriar a temperatura ambiente. Añadir 5 ml de la solución patrón de fosfato que contenga 2 mg de P2O5 y 20 ml de la solución de molibdovanadato modificado. Diluir a 100 y continuar como en 16(b).4.2.
17(b).4.3 Muestras que contienen más del 5 por 100 de P2O5.‒Introducir en un matraz de 100 ml una alícuota de 5 ó 10 ml de tal forma que dicha alícuota contenga de 2 a 5 mg de P2O5 y digerir la muestra como en 17 (b).4.2.
Continuar como en 17(b).4.2, pero sin añadir solución patrón de fosfato.
17(b).5. Cálculo.
17(b).5.1 Preparación de la curva patrón como en 16(b).5.1.
17(b).5.2 Muestras que contienen menos del 5 por 100 de P2O5 [como en 16 (b).5.2].
17(b).5.3 Muestras que contienen más del 5 por 100 de P2O5 [como en 16 (b).5.31.
17(b).6. Referencias.
1. A. O. A. C.: «Official Methods of Analysis». Ed. 1970, página 15/2.046.
22. BORO SOLUBLE EN ÁCIDO
22.1 Principio.
Consiste en pasar el boro a BO3H3, el cual puede ser valorado con un álcali fuerte, previa exaltación de su acidez por formación de un ion complejo con una sustancia polihidroxilada (manitol o sorbitol).
22.2 Material y aparatos.
22.2.1 Balanza analítica.
22.2.2 pH-metro o similar.
22.2.3 Vaso de 250 ml.
22.3 Reactivos.
22.3.1 Solución patrón de ácido bórico.
Disolver 1 g de BO3H3 en H2O y llevar a 1 litro.
1 ml contendrá 0,1748 mg de B.
22.3.2 Solución patrón de NaOH.
Preparar una solución de concentración 0,025 N libre de CO2 y a continuación se titula de la siguiente manera: Pipetear 25 ml de la solución patrón de BO3H3 depositándolos en un Erlenmeyer de 250 ml. Añadir 3 g de ClNa e indicador rojo de metilo, diluyéndose con agua destilada hasta 150 ml; calentar hasta ebullición para expulsar el CO2, enfriar y titular potenciométricamente como en 22.4. Realizar un blanco repitiendo la titulación cambiando los 25 ml de la solución patrón de BO3H3 por 25 ml de agua. Para determinar la equivalencia en B se efectúan los siguientes cálculos:
mg B/ml = 4,369/ml de la solución de NaOH ‒ ml gastados en el blanco.
Esta solución se debe preservar del CO2 atmosférico.
22.3.3 Solución del indicador rojo de metilo.
Disolver 0,1 mg de rojo de metilo en 50 ml de alcohol, diluir hasta 100 ml con agua y filtrar si es necesario.
22.3.4 ClH.
22.3.5 ClH 0,5 N.
22.3.6 ClH 0,02 N.
22.3.7 Solución de (NO3)2 Pb al 10 por 100.
22.3.8 NaOH 0,5 N.
22.3.9 NaOH 0,025 N.
22.3.10 CINa.
22.3.11 Manitol o sorbitol.
22.4 Procedimiento.
Pesar la muestra con exactitud de 1 mg (22.6.1), depositando la cantidad de sustancia pesada en un vaso de 250 ml. Añadir, aproximadamente, 50 ml de H2O y 3 ml de ClH. Calentar a ebullición y mantener ésta durante un tiempo suficiente como para descomponer los carbonatos. Mantener a continuación la solución caliente sin llegar a ebullición durante el procedimiento de eliminación de posibles fosfatos. Añadir unos 10 ml de solución de (NO3)2 Pb al 10 por 100 o 1 ml por cada 1,2 por 100 de P2O5 si el contenido de éste se conoce y es superior al 12 por 100. Añadir CO3H Na hasta neutralizar. Seguidamente añadir unas cuantas gotas de rojo de metilo y continuar añadiendo CO3H Na hasta conseguir un viraje alcalino del rojo de metilo (amarillo o ligeramente anaranjado). A continuación calentar la mezcla al baño de María durante treinta minutos, añadiendo pequeñas cantidades de CO3H Na si fuera necesario para mantener el color (22.6.2). Después de la neutralización y el período de calentamiento, tendremos de 40-50 ml de solución. Filtrar la solución en caliente, recogiéndola sobre un Erlenmeyer de 250 ml, lavando el filtro con agua caliente. Se acidifica con unas cuantas gotas de ClH y calentar brevemente para expulsar el CO2. Neutralizar la solución en caliente con NaOH 0,5 N y acidificar de nuevo con ClH 0,5 N, añadiendo de 0,3-0,5 ml de exceso. Diluir a 150 ml, aproximadamente, y calentar de nuevo a ebullición durante unos minutos para expulsar el CO2 que pueda quedar. Enfriar a temperatura ambiente con agua fría. Neutralizar la mezcla con NaOH 0,5 N y colocar dentro del vaso los electrodos y agitador magnético.
Comenzar a agitar y ajustar a pH = 6,30 mediante la adición de NaOH 0,25 N o ClH 0,02 N y según se requiera (22.6.3). Una vez que el pH se mantenga a 6,30, se procede a efectuar la lectura en la bureta de NaOH 0,25 N; añadir 20 g de manitol o de cristales de D-sorbitol y efectuar la valoración con sosa 0,25 N hasta obtener nuevamente el pH 6,30 (desconectar el pH-metro dejando la lectura a 6,30 cuando se añade el manitol, conectándolo de nuevo cuando el indicador se acerca a su punto final, continuando añadiéndole cuidadosamente solución patrón de NaOH hasta que la aguja del galvanómetro vuelva a cero). De esta manera, con práctica, se trabaja sin sobrepasar el punto final de la titulación. Cuando se alcanza el punto final, se efectúa de nuevo la lectura de la cantidad de NaOH gastado. Efectuar un blanco con todos los reactivos a excepción de la muestra a analizar.
22.5 Cálculos.
Calcular el contenido en B aplicando la siguiente fórmula:
% B = (ml de NaOH gastados en la determinación ‒ ml blanco) ⋅ (mg B/ml de solución de NaOH)/10 g muestra.
22.6 Observaciones.
22.6.1 Para muestras de hasta 0,45 por 100 de contenido en B, pesar 1 g. Para contenidos más altos, pesar menos, pero guardando, aproximadamente, esta proporción.
22.6.2 Si el color de la solución palidece debido a la presencia de nitratos, añadir más rojo de metilo. Por el contrario, si dicho color se oscurece debido a la materia orgánica, se aconseja seguir la neutralización por medio de la técnica del indicador externo.
22.6.3 El pH se debe mantener; de no ocurrir así, sería debido a una expulsión incompleta del CO2.
22.7 Referencias.
1. A. O. A. C.: «Official Methods of Analysis». Ed. 1970, página 24/2.103.
23. BORO SOLUBLE EN AGUA
23.1 Principio.
Consiste en pasar el boro a BO3H3, el cual puede ser valorado con un álcali fuerte, previa exaltación de su acidez por formación de un ión complejo con una sustancia polihidroxilada (manitol o sorbitol).
23.2 Material y aparatos.
23.2.1 Matraces de 250 ml.
23.2.2 Papel de filtro Whatman número 40, o similar.
23.2.3 Matraces de 500 ml.
23.2.4 Titulador automático.
23.3 Reactivos.
23.3.1 Ba(OH)2.
23.3.2 Solución de Cl2Ba al 10 por 100.
23.3.3 Coadyuvante de filtración inerte.
23.3.4 Ácido clorhídrico (1 + 5).
23.4 Procedimiento.
Pesar 2,5 g de muestra y depositarla en un Erlenmeyer de 250 ml. Añadir 125 ml de H2O, calentando a ebullición durante diez minutos, filtrando a continuación a través de papel Whatman número 40, o papel similar, recogiéndose el filtrado sobre un Erlenmeyer de 400 ml. Lavar bien el filtro con seis porciones H2O caliente y diluir hasta 200 ml con H2O. Calentar el filtrado hasta ebullición. Añadir seguidamente 15 ml de solución de Cl2Ba al 10 por 100 con objeto de precipitar los fosfatos y sulfatos. Añadir a continuación Ba(OH)2 en polvo con cuidado y agitando constantemente hasta conseguir un pH alcalino (usando como indicador fenolftaleína) y añadiendo exceso de Ba(OH)2. Calentar a ebullición durante sesenta minutos o más para expulsar el NH3 (muestras que tengan fuerte color debido a la materia orgánica serán mantenidas en ebullición durante un tiempo más prolongado). Si es necesario, añadir agua con objeto de mantener el volumen de 150 ml.
Añadir una o dos cucharadas de un coadyuvante de filtración inerte y filtrar mediante succión a través de pasta de papel prensado, recogiéndolo sobre un Erlenmeyer de 500 ml. Lavar el precipitado con seis porciones H2O hirviendo. Añadir ClH (1 + 5) hasta decolorar la fenolftaleína que contenía el filtrado. Añadir rojo de metilo y, mediante sucesivas adiciones de ClH, llevarlo a color rosa. Añadir plato poroso y el agitador, cubrir con vidrio de reloj y calentar durante cinco minutos a ebullición para expulsar el CO2. Enfriar con agua teniendo la precaución de mantenerlo cubierto con el vidrio de reloj. Lavar agitador, vidrio de reloj y Erlenmeyer comenzando la titulación añadiendo solución patrón de NaOH 0,05 N hasta color amarillo del rojo de metilo (23.3.2). Añadir 20 g de D-manitol y 1 ml o más de fenolftaleína, agitar y lavar las paredes del Erlenmeyer. Efectuar la titulación hasta viraje rosa del indicador. Efectuar un blanco repitiendo todas las operaciones efectuadas con la muestra.
23.5 Cálculos.
Calcular el contenido en B aplicando la siguiente fórmula:
1 ml de NaOH 0,05 N = 0,000540 g de B = 0,00477 g Na2B4O7 ⋅ 10 H2O
23.6. Referencias.
1. A. O. A. C.: «Official Methods of Analysis». Ed. 1970, página 25/2016.
24. DETERMINACIÓN DEL MAGNESIO
(Por absorción atómica)
24.1 Principio.
Consiste en solubilizar el Mg de la muestra con diferentes ácidos, según el tipo de fertilizante, y se mide su absorción a 285,2 nm, refiriéndola a la correspondiente curva patrón.
24.2 Material y aparatos.
24.2.1 Matraces aforados de 50, 100 y 1.000 ml de capacidad.
24.2.2 Espectrofotómetro de absorción atómica.
Longitud de onda: 285,2 nm.
Gases: aire-C2H2.
Rango: 0,2-2 μg/ml.
24.3 Reactivos.
24.3.1 Solución patrón de Mg 1.000 μg/ml.‒Pesar 1.000 mg de metal puro Mg, agregar 50 ml de agua y añadir lentamente 10 ml de ClH. Diluir a un litro.
24.3.2 Solución de lantano al 5 por 100.‒50 g de La por litro en ClH, aproximadamente, del 5 por 100. Disolver 58,65 g de La2O3 en 250 ml de ClH (1:1) diluyendo a un litro con agua destilada.
24.3.3 ClH.
24.3.4 ClH 2 N.
24.3.5 ClH 0,5 N.
24.3.6 AlH (1:1).
24.3.7 CIO4H al 70 por 100 d = 1,67.
24.3.8 Ácido fluorhídrico.
24.3.9 Metanol.
24.4 Procedimiento.
24.4.1 Materiales inorgánicos y mezcla de fertilizantes.
Disolver 1 g de muestra muy bien homogeneizada e introducir en un Erlenmeyer de 150 ml; añadir 10 ml de ClH concentrado. Evaporar la solución a casi sequedad por ebullición. Redisolver el residuo en 20 ml de ClH 2 N, hirviéndolo para su disolución si es necesario. Filtrar a través de papel rápido recogiéndolo en un matraz de 100 ml, lavando el papel de filtro y el Erlenmeyer con agua. Medir la absorbancia de esta solución directamente o diluir con ClH 0,5 N para obtener soluciones que entren dentro del rango del aparato.
24.4.2 Fertilizantes que contengan materia orgánica.‒Pesar 1 g de muestra y ponerla en una cápsula de porcelana; calentar en la mufla a 500 °C durante una hora. Añadir seguidamente 10 ml de ClH y continuar como en 24.4.1.
24.4.3 Fertilizantes que contengan elementos fritados.
Disolver 1 g, o menos, de muestra bien pulverizada en 5 ml de CIO4H y 5 ml de FH. Evaporar a ebullición hasta desaparición de los humos densos del CIO4H. Diluir cuidadosamente con H2O, filtrar y proceder como en 24.4.1. Alternativamente se disuelve la muestra en 10 ml de ClH, 5 ml de FH y 10 ml de metanol. Evaporar a sequedad. Añadir 5 ml de ClH y volver a evaporar. Repetir la adición de ClH y la subsiguiente evaporación. Disolver el residuo como en 24.4.1.
24.4.4 Determinación.‒(El P puede interferir al Mg con llama de aire C2H2. Se elimina la interferencia por adición de la solución de lantano a las soluciones preparadas a partir de la muestra y a las soluciones patrones, de manera que las diluciones finales contengan, al menos, 1 por 100 de La).
Ajustar las condiciones óptimas del aparato. Se puede utilizar para evitar diluciones una línea secundaria menos sensible. Leer, por lo menos, cuatro puntos de la curva de calibrado después de medir cada grupo de 6-12 muestras.
Restablecer el cero con agua cada vez que sea necesario. Se debe efectuar una curva de calibrado antes y después de efectuar la medida de cada grupo de muestras. Dibujándose la curva de calibrado con absorciones contra concentraciones expresadas en μg/ml.
24.5 Cálculos.
Porcentaje de elemento = (μg/ml) x (F/peso de la muestra • 104)
Siendo F = ml de la dilución original.
x ml de la dilución final/ml de la alícuota tomada.
Esta fórmula se utilizará cuando el volumen original de 100 ml se diluya.
24.6 Referencias.
1. A. O A. C.: «Official Methods of Analysis». Ed. 1970, página 24/2.097.
25. AZUFRE LIBRE
25.1 Principio.
Extracción del azufre con sulfuro de carbono, oxidación del azufre o sulfato y posterior precipitación del sulfato mediante cloruro bárico.
25.2 Material y aparatos.
25.2.1 Vaso de 250 ml.
25.2.2 Extractor tipo Soxhlet.
25.2.3 Horno eléctrico regulable.
25.2.4 Baño de agua.
25.2.5 Papel Whatman número 42 o similar o filtro Gooch.
25.3 Reactivos.
25.3.1 Sulfuro de carbono.
25.3.2 Solución saturada de bromo en tetracloruro de carbono.
25.3.3 Ácido nítrico concentrado.
25.3.4 Ácido clorhídrico concentrado.
25.3.5 Ácido clorhídrico al 2 por 100.
25.3.6 Solución de azul de bromofenol.‒Disolver 0,1 g de azul de bromofenol en 1,5 ml de hidróxido sódico 0,1 N y diluir a 25 ml con agua destilada.
25.3.7 Hidróxido amónico concentrado.
25.3.8 Solución de cloruro de bario al 10 por 100.‒Disolver 100 g de Ba Cl2.2H2O en 900 ml de agua destilada. Filtrar a través de papel Whatman número 42 o equivalente.
25.4 Procedimiento.
Extraer 1 g de muestra con sulfuro de carbono en el extractor Soxhlet y dejar drenar el cartucho al menos doce veces. Transferir el extracto a un vaso de 250 ml y evaporar el sulfuro de carbono en corriente de aire a temperatura ambiente. Calentar en estufa durante veinte minutos a 60-70 °C y dejar enfriar a temperatura ambiente; una vez frío, añadir 10 ml de la solución 25.3.2 y dejar tapado durante veinte minutos, agitando varias veces. Añadir 15 ml de ácido nítrico y dejar tapado de nuevo durante treinta minutos, volviendo a agitar suavemente, Evaporar en baño de agua hirviendo hasta reducir el volumen aproximadamente a 5 ml, añadir 20 ml de ácido clorhídrico y evaporar igualmente hasta reducir el volumen a 5 ml. Añadir 50 ml de agua, filtrar y lavar el filtro con ácido clorhídrico al 2 por 100. Añadir al filtrado dos gotas de azul de bromofenol y después hidróxido amónico hasta el primer cambio de color. Añadir lentamente ácido clorhídrico hasta reacción netamente ácida más cinco gotas en exceso; diluir a 150 ml con agua, calentar a ebullición y añadir lentamente gota a gota la solución 25.3.8 aproximadamente hasta el 50 por 100 de exceso. Tapar el vaso y ponerlo al baño de agua hirviendo durante una hora como mínimo, enfriar a la temperatura ambiente y filtrar a través de papel de filtro Whatman número 42 o equivalente o de un filtro Gooch previamente desecado por calcinación a 800 °C, al menos, durante veinte minutos. Lavar diez veces el filtro con agua caliente, desecar y calcinar a 800 °C hasta peso constante.
25.5 Cálculo.
Calcular el contenido del azufre en tanto por ciento.
P = peso, en g, de las cenizas.
P’ = peso, en g, de la muestra.

25.6 Referencias.
1. Association of Official Agricultural Chemists. «Official Methods of Analysis», 1975, 2.149, pág. 29.
26. AZUFRE TOTAL
26.1 Principio.
Oxidación del azufre o sulfato y posterior precipitación del sulfato con cloruro bárico.
26.2 Material y aparatos.
26.2.1 Vaso de 250 ml.
26.2.2 Papel Whatman número 42 o similar o filtro Gooch.
26.2.3 Baño de agua.
26.2.4 Horno eléctrico regulable.
26.3 Reactivos.
26.3.1 Solución de cloruro de bario al 10 por 100.‒Disolver 100 g de Ba Cl2.2H2O en 900 ml de agua destilada. Filtrar a través de papel Whatman número 42 o equivalente,
26.3.2 Solución de bromo al 10 por 100.‒Disolver 10 g de bromo en 90 g de tetracloruro de carbono y agitar hasta homogeneidad. Guardar en frasco con tapón de vidrio.
26.3.3 Ácido nítrico concentrado.
26.3.4 Ácido clorhídrico concentrado.
26.3.5 Solución de nitrato de plata al 1 por 100.
26.4 Procedimiento.
26.4.1 Preparación de la muestra.‒Pesar una cantidad de muestra que contenga entre 50-150 mg de azufre y colocarla dentro de un vaso de 250 ml de capacidad. A continuación añadir 20 ml de la solución 26.3.2. Mezclar, agitando el vaso a intervalos de cinco minutos, durante media hora. Añadir 15 ml de ácido nítrico y mezclar de la misma forma que anteriormente. Evaporar en baño de agua hirviendo hasta reducir el volumen a 1-2 ml. Añadir 15 ml de ácido clorhídrico y 10 ml de H2O y evaporar hasta sequedad en baño de agua. Añadir 10 ml de ácido clorhídrico y 50 ml de H2O, calentarlo hasta ebullición y hervir durante cinco minutos, a continuación filtrar a través de papel Whatman número 42 o equivalente. Lavar el papel con 20 ml de agua caliente, efectuando esta operación diez veces.
26.4.2 Determinación.‒A partir del filtrado obtenido según 26.4.1, calentar hasta ebullición y añadir 5-6 gotas de la solución 26.3.1. Pasado un minuto, añadir lentamente una cantidad de la solución 26.3.1 equivalente al contenido supuesto de azufre (1 ml = 0,014 g de S), más 5 ml en exceso. Llevar a ebullición suave durante una hora: pasado este tiempo, retirar y dejar reposar durante quince-veinte minutos hasta la sedimentación del precipitado, a continuación filtrar a través de un filtro de Gooch, previamente desecado por ignición y tarado, o papel Whatman número 42 o equivalente. Lavar con agua caliente hasta que 10 ml del filtrado no precipite al añadir 3 ml de nitrato de plata al 1 por 100. A continuación desecar e incinerar a 800 °C hasta peso constante.
26.5 Cálculo.
Calcular el contenido en azufre en tanto por ciento.
P = peso, en g, de las cenizas.
P’ = peso, en g, de la muestra.
26.6 Referencias.
1. Association of Official Agricultural Chemists. «Official Methods of Analysis». Ed. 1975, 2.148, pág. 39.
27. AZUFRE DE SULFATOS
27.1 Principio.
Solubilización del sulfato en medio ácido, y precipitación con cloruro bárico. Aplicación a fertilizantes compuestos, sulfato potásico y sulfato magnésico.
27.2 Material y aparatos.
27.2.1 Vasos de 250 ml.
27.2.2 Papel Whatman número 42, o similar, o filtro Gooch.
27.2.3 Baño de agua.
27.2.4 Horno eléctrico regulable.
27.3 Reactivos.
27.3.1 Solución de cloruro de bario al 10 por 100.‒Disolver 100 g de Ba Cl2.2H2O en 900 ml de agua destilada. Filtrar a través de papel Albet número 242 o equivalente.
27.3.2 Ácido clorhídrico concentrado.
27.3.3 Solución de nitrato de plata al 1 por 100.
27.4 Procedimiento.
27.4.1 Preparación de la muestra.‒Pesar con exactitud de 0,1 mg una cantidad de muestra que contenga 0,1 a 0,2 g de SO4, pasar a un vaso de 250 ml y añadir 25 ml de ácido clorhídrico concentrado y 50 ml de H2O. Hervir durante diez minutos.
A continuación filtrar a través de papel Whatman número 42 o equivalente. Lavar con unas diez porciones de 10 ml de agua caliente.
27.4.2 Determinación.‒El filtrado obtenido se calienta hasta ebullición y se añaden cinco-seis gotas de solución 27.3.1. Pasado un minuto añadir lentamente una cantidad de solución 27.3.1, equivalente al contenido supuesto de azufre (1 ml =0,042 g de SO4=) más 5 ml en exceso. Llevar a ebullición suave durante una hora, pasado este tiempo retirar y dejar reposar durante quince-veinte minutos hasta la sedimentación del precipitado, a continuación filtrar a través de un filtro Gooch, previamente desecado por calcinación y tarado, o papel Albet número 242 o equivalente. Lavar con agua caliente hasta que 10 ml de filtrado no precipiten al añadirle 3 ml de la solución 27.3.3. A continuación, desecar y calcinar a 800 °C hasta peso constante.
27.5 Cálculo.
Calcular el contenido en azufre o sulfato en tanto por ciento.
P = peso, en g, de las cenizas.
P’ = peso, en g, de la muestra.


27.6 Referencias.
1. Association of Official Agricultural Chemists. «Official Methods of Analysis». Ed. 1975, 2.148, pág. 29.
28. CLORUROS
(Solubles en agua)
28.1 Principio.
Solubilización de los cloruros en agua y subsiguiente valoración con nitrato de plata en presencia de cromato potásico.
28.2 Material y aparatos.
28.2.1 Material necesario para filtración.
28.2.2 Matraz aforado de 250 ml de capacidad.
28.2.3 Vaso de precipitados de 150 ml de capacidad.
28.2.4 Material necesario para volumetría.
28.3 Reactivos.
28.3.1 Solución de nitrato de plata.‒Disolver 5 g de nitrato de plata recristalizado y enrasar con agua a 1.000 ml. Titular dicha solución con cloruro sódico, de manera que 1 ml de la solución de nitrato de plata equivalga a 0,001 g de cloro.
28.3.2 Indicador de cromato potásico al 5 por 100 (p/v).‒Disolver 5 g de cromato potásico en 100 ml de agua.
28.4 Procedimiento.
Pesar 2,5 g de muestra y ponerla sobre un papel de filtro de 11 cm, lavar a continuación con sucesivas porciones de agua hirviendo hasta su completo lavado; aproximadamente se emplean unos 250 ml de agua.
Recoger el filtrado sobre un matraz aforado de 250 ml, enfriar y enrasar con agua.
Tomar 50 ml de filtrado y llevarlos a un vaso de precipitados de 150 ml de capacidad, añadir 1 ml indicador de cromato potásico al 5 por 100 (p/v), ajustar el pH entre 6 y 7 y valorar con la solución de nitrato de plata (28.3.1.) hasta color rojo constante de cromato de plata.
28.5 Cálculos.
Calcular el contenido en cloro expresado en gramos:
Cl‒ en g = V 0,001.
Siendo:
V = volumen en ml gastados de la solución de nitrato de plata.
28.6 Referencias.
1. Association of Official Analytical Chemists. «Official Methods of Analysis». Ed. 1975. 39, pág. 24.
29. CINC
29.1 Principio.
Solubilizar el Zn de la muestra con diferentes ácidos, según el tipo de fertilizante, y medir su absorción a 213,8 nm, refiriéndola a la correspondiente curva patrón.
29.2 Material y aparatos.
29.2.1 Vaso de precipitado de 150 ml de capacidad.
29.2.2 Matraces aforados de 100 y 1.000 ml de capacidad.
29.2.3 Espectrofotómetro de absorción atómica.
Longitud de onda: 213,8 nm.
Gases: aire-acetileno.
Rango: 0,5-5 ppm.
29.2.4 Cápsula o crisol de platino.
29.2.5 Cápsula o crisol de porcelana.
29.3 Reactivos.
29.3.1 Solución patrón de cinc de 1.000 ppm.‒Disolver en 10 ml de ácido clorhídrico 6 N, 1.000 g de cinc metal y llevar a 1.000 ml con agua destilada.
29.3.2 Ácido clorhídrico.
29.3.3 Ácido clorhídrico 6 N.
29.3.4 Ácido clorhídrico 2 N.
29.3.5 Ácido clorhídrico 0,5 N.
29.3.6 Ácido fluorhídrico.
29.3.7 Ácido perclórico.
29.3.8 Metanol.
29.4 Procedimiento.
29.4.1 Materiales inorgánicos y fertilizantes compuestos.‒ Disolver 1 g de muestra bien molida en 10 ml de ácido clorhídrico utilizando un vaso de precipitado de 150 ml. Someter a ebullición hasta casi sequedad, sin dejar que la evaporación sea completa. Redisolver el residuo en 20 ml de ácido clorhídrico 2 N calentando a ebullición si es necesario. Filtrar, a través de papel de filtrado rápido, sobre un matraz de 100 ml, lavando completamente el papel y el residuo con agua destilada. Medir la absorción de la disolución directamente, o en caso necesario diluir con ácido clorhídrico 0,5 N hasta conseguir una solución dentro del rango de la curva de calibrado.
29.4.2 Fertilizantes conteniendo materia orgánica.‒Pesar 1 g de muestra en una cápsula de porcelana y calentar en una mufla en corriente de aire durante una hora a 500 °C; romper la pasta y disolver en 10 ml de ácido clorhídrico como en 29.4.1.
29.4.3 Fertilizantes fritados que contengan microelementos.‒Disolver 1 g de muestra bien molida en 5 ml de ácido perclórico y 5 ml de ácido fluorhídrico. Calentar a ebullición hasta aparición de humos densos de ácido perclórico, diluir cuidadosamente con agua destilada, filtrar y proceder como en 29.4.1.
Alternativamente también se puede disolver la muestra en 10 ml de ácido clorhídrico, 5 ml de ácido fluorhídrico y 10 ml de metanol. Evaporar a sequedad. Añadir 5 ml de ácido clorhídrico y evaporar. Repetir la adición de ácido clorhídrico y la evaporación. Disolver el residuo como en 29.4.1.
29.5 Cálculos.

Siendo:
L = valor, en ppm, obtenido en la curva patrón.
P = peso, en g, de la muestra.
F = ml de la dilución original x ml de la dilución final/ml de alícuota. Este factor se aplicará en caso de que se diluya el volumen original de 100 ml.
29.6 Referencias.
1. Association of Official Agricultural Chemists. Ed. 1975, páginas 22-23.
30. COBRE
30.1 Principio.
Solubilizar el Cu de la muestra con diferentes ácidos, según el tipo de fertilizante, y medir su absorción a 324,7 nm refiriéndola a la correspondiente curva patrón.
30.2 Material y aparatos.
30.2.1 Vaso de precipitado de 150 ml de capacidad.
30.2.2 Matraces aforados de 100 y 1.000 ml de capacidad.
30.2.3 Espectrofotómetro de absorción atómica.
Longitud de onda: 324,7 nm.
Gases: aire-acetileno.
Rango: 2-20 ppm.
30.2.4 Cápsula o crisol de platino.
30.2.5 Cápsula o crisol de porcelana.
30.3 Reactivos.
30.3.1 Solución patrón de cobre de 1.000 ppm.‒Disolver en la mínima cantidad posible de ácido nítrico concentrado 1,000 g de cobre metal y añadir 5 ml de ácido clorhídrico concentrado. Evaporar sin llegar a total sequedad y diluir a 1.000 ml con ácido clorhídrico 0,1 N.
30.3.2 Ácido clorhídrico.
30.3.3 Ácido nítrico.
30.3.4 Ácido clorhídrico 2 N.
30.3.5 Ácido clorhídrico 0,1 N.
30.3.6 Ácido clorhídrico 0,5 N.
30.3.7 Ácido fluorhídrico.
30.3.8 Ácido perclórico.
30.3.9 Metanol.
30.4 Procedimiento.
30.4.1 Materiales inorgánicos y fertilizantes compuestos (como en 29.4.1).
30.4.2 Fertilizantes conteniendo materia orgánica (como en 29.4.2).
30.4.3 Fertilizantes fritados que contengan microelementos (como en 29.4.3).
30.5 Cálculos.

Siendo:
L = ppm leídos en la curva patrón.
P = peso, en g, de la muestra.
F = mi de la dilución original x ml de la dilución final/ml de alícuota. Este factor se aplicará en caso de que se diluya el volumen original de 100 ml.
30.6 Referencias.
1. Association of Official Agricultural Chemists. 49 Ed. 1975, páginas 22-23.
31. SODIO
31.1 Principio.
Determinación del sodio por fotometría de llama.
31.2 Material y aparatos.
31.2.1 Fotómetro de llama y accesorios.
31.2.2 Matraces aforados de 100, 250, 500 y 1.000 ml de capacidad.
31.3 Reactivos.
31.3.1 Solución de oxalato amónico.‒Añadir a 40 g de oxalato amónico agua destilada hasta 1.000 ml y disolver.
31.3.2 Indicador rojo de metilo.‒Disolver 0,2 g de rojo de metilo en 100 ml de alcohol.
31.3.3 Cloruro sódico.‒Desecar durante dos horas a 105 °C.
31.3.4 Ácido nítrico al 1 por 100 (v/v).
31.4 Procedimiento.
31.4.1 Preparación de la solución de la muestra de fertilizantes compuestos sulfato magnésico y sulfato potásico.
Pesar 2,5 g de muestra (para contenidos menores del 4 por 100 en sodio) o 1,25 g (para contenidos entre 4-20 por 100). Introducirla en un matraz aforado de 150 ml de capacidad, añadir 125 ml de agua y 50 ml de la solución 31.3.1; llevar a continuación a ebullición durante treinta minutos, dejar enfriar y enrasar con agua, agitar y filtrar. Transferir a continuación 25 ml (para contenidos menores del 4 por 100 en sodio) o 10 ml (para contenidos comprendidos entre 4 y 20 por 100 en sodio) de dicha solución a un matraz aforado de 250 ml de capacidad, enrasar con agua y agitar. Medir a continuación refiriendo la lectura a la curva patrón 31.4.2.1 ó 31.4.2.2, representadas en emisión y concentraciones.
31.4.2 Preparación de la curva patrón.
31.4.2.1 Solución patrón para muestras con contenidos igual o superiores al 1 por 100 en sodio.‒Disolver 1,2716 g de cloruro sódico en agua y enrasar a 500 ml; esta solución corresponde a 1.000 ppm de sodio. Tomar alícuotas de la solución patrón de manera que cubran un rango comprendido entre 0 y 40 ppm con intervalos de 5 ppm.
31.4.2.2 Solución patrón para muestras con contenidos inferiores al 1 por 100 de sodio.‒Proceder como en 31.4.2.1, pero cubriendo un rango comprendido entre 0 y 10 ppm con intervalos de 2 ppm.
31.5 Cálculo.
Calcular el contenido en tanto por ciento de sodio.
31.5.1 Para muestras con contenidos comprendidos entre 0 y 4 por 100 en sodio.

A = valor obtenido en la curva patrón expresado en ppm.
31.5.2 Para muestras con contenidos comprendidos entre 4 y 20 por 100 en sodio.

B = valor obtenido en la curva patrón expresado en ppm.
31.6 Referencias.
1. Association of Official Agricultural Chemists. «Official Methods of Analysis». 1975 50. 2.134, pág. 29.
32. HIERRO
32.1 Principio.
Solubilizar el Fe de la muestra con diferentes ácidos, según el tipo de fertilizante, y medir su absorción a 248,3 nm refiriéndola a la correspondiente curva patrón.
32.2 Material y aparatos.
32.2.1 Vaso de precipitado de 150 ml de capacidad.
32.2.2 Matraz aforado de 100 ml de capacidad.
32.2.3 Espectrofotómetro de absorción atómica.
Longitud de onda: 248,3 nm.
Gases: aire-acetileno.
Rango: 2-20 ppm.
32.2.4 Cápsula o crisol de platino.
32.2.5 Cápsula o crisol de porcelana.
32.3 Reactivos.
32.3.1 Solución patrón de Fe de 1.000 ppm ‒Disolver calentando a ebullición 1.000 g de Fe puro en 30 ml de ClH N, agregado en sucesivas proporciones. Diluir hasta 1.000 ml con H2O destilada. Diluir con ClH 0,5 N preparando cuatro patrones, al menos, centro del rango de detección.
32.3.2 Ácido clorhídrico.
32.2.3 Ácido clorhídrico 6 N.
32.3.4 Ácido clorhídrico 2 N.
32.3.5 Ácido clorhídrico 0,5 N.
32.3.6 Ácido fluorhídrico.
32.3.7 Ácido perclórico.
32.3.8 Metanol.
32.4 Procedimiento.
32.4.1 Materiales inorgánicos y fertilizantes compuestos (como en 29.4.1).
32.4.2 Fertilizantes conteniendo materia orgánica (como en 29.4.2).
32.4.3 Fertilizantes fritados que contengan microelementos (como en 29.4.3).
32.5 Cálculos.

Siendo:
L = ppm leídos en la curva patrón.
P = peso, en g, de la muestra.
F = ml de la dilución original x ml de la dilución final/ml de alícuota. Este factor se aplicará en caso de que se diluya el volumen original de 100 ml.
32.6 Referencias.
1. Association of Official Agricultural Chemists. «Official Methods of Analysis». Ed. 1975. 42, págs. 22-23.
33. MANGANESO
33.1 Principio.
Solubilizar el Mn de la muestra con diferentes ácidos, según el tipo de fertilizante, y medir su absorción a 279,5 nm, refiriéndola a la correspondiente curva patrón.
33.2 Material y aparatos.
33.2.1 Vaso de precipitado de 150 ml de capacidad.
33.2.2 Matraces aforados de 100 y 1.000 ml de capacidad.
33.2.3 Espectrofotómetro de absorción atómica.
Longitud de onda: 279,5 nm.
Gases: aire-acetileno.
Rango: 2-20 ppm.
33.2.4 Cápsula o crisol de platino.
33.2.5 Cápsula o crisol de porcelana.
33.3 Reactivos.
33.3.1 Solución patrón de manganeso de 1.000 ppm.‒Disolver en 30 ml de ácido clorhídrico 6 N, 1,582 g de bióxido de manganeso, llevar a ebullición hasta evaporación del ácido clorhídrico y enrasar a 1.000 ml con agua destilada.
33.3.2 Ácido clorhídrico.
33.3.3 Ácido clorhídrico 6 N.
33.3.4 Ácido clorhídrico 2 N.
33.3.5 Ácido clorhídrico 0,5 N.
33.3.6 Ácido fluorhídrico.
33.3.7 Ácido perclórico.
33.3.8 Metanol.
33.4 Procedimiento.
33.4.1 Materiales inorgánicos y fertilizantes compuestos (como en 29.4.1).
33.4.2 Fertilizantes conteniendo materia orgánica (como en 29.4.2).
33.4.3 Fertilizantes fritados que contengan microelementos (como en 29.4.3).
33.5 Cálculos.

Siendo:
L = ppm leídos en la curva patrón.
P = peso, en g, de la muestra.
F = ml de la dilución original x ml de la dilución final/ml de alícuota. Este factor se aplicará en caso de que se diluya el volumen original de 100 ml.
33.6 Referencias.
1. Association of Official Agricultural Chemists. «Official Methods of Analysis». Ed. 1975. 42, págs. 22-23.
34(a). CALCIO SOLUBLE EN ÁCIDO
(Volumetría)
34(a).1 Principio.
Se determina el calcio soluble en ácido nítrico y clorhídrico, mediante una valoración del oxalato cálcico, formado con permanganato potásico.
Aplicable a concentraciones superiores al 0,1 por 100.
34(a).2 Material y aparatos.
34(a).2.1 Matraces aforados de 250 ml.
34(a).2.2 Vasos de precipitado de 250 ml.
34(a).2.3 Papel de filtro cuantitativo o crisoles Gooch.
34(a).2.4 Bureta de 25 ml.
34(a).3 Reactivos.
34(a).3.1 Ácido nítrico.
34(a).3.2 Ácido clorhídrico.
34(a).3.3 Solución indicador azul de bromofenol.‒Mezclar 0,1 g de azul de bromofenol con 1,5 ml de hidróxido sódico 0,1 N y diluir a 25 ml.
34(a).3.4 Amoníaco diluido (1:4) (v/v).
34(a).3.5 Ácido clorhídrico diluido (1:4) (v/v).
34(a).3.6 Solución saturada de oxalato amónico.
34(a).3.7 Ácido sulfúrico.
34(a).3.8 Solución valorada de permanganato potásico 0,1 N.
34(a).3.9 Amoníaco diluido (1:50) (v/v).
34(a).4 Procedimiento.
Pesar 2,50 g de muestra y pasarlos a un matraz aforado de 250 ml, añadir 30 ml de ácido nítrico y 10 ml de ácido clorhídrico y hervir treinta minutos. Enfriar, diluir, aforar y filtrar si es necesario. Llevar 25 ml de la solución a un vaso y diluir a 100 ml. Añadir dos gotas de azul de bromofenol. Añadir solución de amoníaco diluido (1:4) hasta que el indicador cambie del amarillo al verde (no al azul). Si se ha sobrepasado este punto, retroceder con clorhídrico (1:4) (pH 3,5 a 4,0). Diluir a 150 ml, llevar a ebullición y añadir 30 ml de solución saturada caliente de oxalato amónico, lentamente y con agitación constante. Si el color cambia del verde al azul o al amarillo de nuevo, ajustar al verde con ácido clorhídrico (1:4). Si pasa al amarillo, ajustar con amoníaco diluido (1/4) al verde. Digerir en baño de agua durante una hora, o dejar toda la noche y enfriar a temperatura ambiente. Filtrar el líquido sobrenadante a través del papel cuantitativo o crisol Gooch o filtro de vidrio fritado y lavar el precipitado con amoníaco diluido (1:50).
Colocar el papel o el crisol con el precipitado, en el vaso original y añadir una mezcla de 125 ml de agua y 5 ml de ácido sulfúrico. Calentar a unos 70 °C y valorar con solución de permanganato potásico 0,1 N, hasta persistencia de color rosa débil. Corregir con un blanco y calcular como calcio.
34(a).5 Cálculo.

Siendo:
V = volumen, en ml, de la solución de permanganato potásico consumidos.
N = normalidad del permanganato potásico.
P = peso, en g, de muestra.
34(a).6 Referencias.
1. Association of Official Agricultural Chemists. «Official Methods of Analysis». 1975. 40. pág. 24.
34(b). CALCIO SOLUBLE EN ÁCIDO
(Absorción atómica)
34(b).1 Principio.
Se determina el calcio soluble en ácido, por absorción atómica en disoluciones al 1 por 100 de lantano y ácido clorhídrico 0,5 N.
34(b).2 Material y aparatos.
34(b).2.1 Vasos de precipitado de 150 ml.
34(b).2.2 Matraces aforados de 25, 250 y 1.000 ml.
34(b).2.3 Espectrofotómetro de absorción atómica.
Longitud de onda: 422,7 nm.
Gases: aire-acetileno.
Rango: 2-20 ppm.
34(b).2.4 Cápsula o crisol de platino.
34(b).2.5 Cápsula o crisol de porcelana.
34(b).3 Reactivos.
34(b).3.1 Solución patrón de 25 ppm de calcio.‒Disolver 1,249 g de carbonato cálcico en la mínima cantidad de ácido clorhídrico 3 N. Diluir a 1 litro. Tomar 50 ml de esta solución y diluir a 1 litro.
34(b).3.2 Solución de lantano (50 g La/l).‒Disolver 58,65 gramos de óxido de lantano en 250 ml de ácido clorhídrico, añadiendo el ácido lentamente. Diluir a 1 litro.
34(b).3.3 Ácido nítrico.
34(b).3.4 Ácido clorhídrico.
34(b).3.5 Ácido clorhídrico 0,5 N.
34(b).3.6 Ácido clorhídrico 2 N.
34(b).3.7 Ácido fluorhídrico.
34(b).3.8 Ácido perclórico.
34(b).3.9 Metanol.
34(b).4 Procedimiento.
34(b).4.1 Materiales inorgánicos y fertilizantes compuestos. Pesar 2,50 g de muestra en un matraz aforado de 250 ml, añadir 30 ml de ácido nítrico y 10 ml de ácido clorhídrico y hervir durante treinta minutos. Enfriar, diluir a volumen, mezclar y filtrar si es necesario. Medir la absorción de la disolución filtrada, previamente diluida con ácido clorhídrico 0,5 N, hasta conseguir una solución dentro del rango de la curva de calibrado, habiendo añadido la cantidad de solución de lantano para hacer la solución final al 1 por 100 en lantano.
34(b).4.2 Fertilizantes conteniendo materia orgánica.‒Como en 29.4.2, pero añadiendo la cantidad de solución de lantano necesaria para obtener una solución final al 1 por 100.
34(b).4.3 Fertilizantes fritados que contengan microelementos.‒Como en 29.4.3, pero añadiendo lantano como se indica en el párrafo anterior.
34(b).4.4 Preparación de la curva patrón.‒Tomar 0,5, 10, 15 y 20 ml de la solución patrón de calcio [34 (b).3.1] y pasarlos a matraces aforados de 25 ml. Añadir a cada uno 5 ml de solución patrón de lantano [34 (b).3.2] y diluir a 25 ml. Estas soluciones contienen, respectivamente, 0,5, 10, 15 y 20 ppm de Ca y 1 por 100 de La.
34(b).5 Cálculos.

Siendo:
L = valer en ppm obtenido en la curva patrón.

P = peso, en g, de muestra.
34(b).6 Referencias.
1. Association of Official Agricultural Chemists. «Official Methods of Analysis» 1975. 40, pág. 24.
MÉTODOS FÍSICOS
1. ESTABILIDAD DE UNA EMULSIÓN
(Dilución 5 por 100 v/v)
1.1 Principio.
La estabilidad de una emulsión obtenida dispersando el concentrado emulsionable en agua patrón se estima por el volumen de aceite o de crema que se separa cuando se mantiene la emulsión en reposo durante veinticuatro horas. También se determina la capacidad de reemulsión del sistema al cabo de veinticuatro horas.
El procedimiento descrito en este método es de aplicación a cualquier concentrado emulsionable que dé lugar al diluirlo con agua a emulsiones del tipo aceite en agua.
1.2 Material y aparatos.
1.2.1 Probeta graduada de 100 ml con tapón de vidrio. El volumen entre la marca de los 100 ml y el fondo del tapón no debe ser mayor de 40 ml ni menor de 35 ml (1.5.1).
1.2.2 Baño de agua de dimensiones adecuadas para permitir la inmersión hasta el cuello en posición vertical, de varias probetas graduadas de 100 ml y con temperatura mantenida a 30 ± 1 °C (1.5.2).
1.2.3 Lámpara ajustable (1.5.6).
1.2.4 Probetas graduadas de 5 ml.
1.3 Reactivos.
1.3.1 Agua patrón.‒342 ppm de dureza pH = 6 a 7 Ca/Mg = 80/20 (p/p) (3.6.1).
1.3.1.1 Carbonato cálcico, con una riqueza no menor del 99 por 100; calentar durante dos horas a 400 °C antes de pesar.
1.3.1.2 Óxido de magnesio, con una riqueza no menor del 99 por 100. Secar durante dos horas a 105 °C antes de pesar.
1.3.1.3 Solución amoniacal, aproximadamente 1 N.
1.3.1.4 Ácido clorhídrico, soluciones 0,1 N y 1 N.
1.3.1.5 Hidróxido sódico 0,1 N.
1.3.1.6 Rojo de metilo, al 0,1 por 100.
1.3.1.7 Solución A.‒Solución de ión cálcico 0,04 M.
Pesar con exactitud 4 g de carbonato cálcico y transferirlo a un Erlenmeyer de 500 ml con un mínimo de agua destilada. Colocar un pequeño embudo en la boca del frasco. Añadir lentamente ácido clorhídrico (82 ml de solución 1,0 N) al frasco, a través del embudo y remover el contenido.
Cuando se disuelva todo el carbonato cálcico, diluir la solución aproximadamente a 400 ml con agua destilada y hervir para eliminar el exceso de dióxido de carbono. Enfriar la solución, añadir indicador rojo de metilo (dos gotas) y neutralizar hasta color naranja con la solución amoniacal añadida gota a gota. Transferir cuantitativamente a un matraz aforado de 1.000 ml, enrasar con agua destilada, mezclar y almacenar en un frasco de polietileno. Un ml de esta solución cuando se diluye hasta 1.000 ml, da un agua que contiene 4 ppm de dureza, expresada como carbonato cálcico.
1.3.1.8 Solución B.‒Solución de ión magnesio 0,04 M.
Pesar con exactitud óxido de magnesio (1,613 g) y transferir a un Erlenmeyer con un mínimo de agua destilada. Poner un pequeño embudo en la boca del Erlenmeyer y añadir lentamente ácido clorhídrico (82 ml de una solución 1N).
Calentar, lentamente, hasta disolver, y cuando todo el óxido de magnesio esté en solución diluir aproximadamente a 400 ml con agua destilada y hervir para eliminar el dióxido de carbono.
Enfriar, añadir una solución de rojo de metilo (dos gotas) y neutralizar hasta color naranja con la solución amoniacal.
Transferir cuantitativamente a un matraz aforado de 1.000 ml, enrasar con agua destilada, mezclar y almacenar en un frasco de polietileno. Un ml de esta solución, cuando se diluye hasta 1.000 ml, da un agua que contiene 4 ppm de dureza, expresada como carbonato cálcico.
1.3.1.9 Preparación‒Llevar 68,5 ml de solución A y 17,0 ml de solución B a un vaso de precipitados de 1.000 ml y diluir a 800 ml, aproximadamente, con agua desionizada. Usando un pH-metro, ajustar la solución a pH 6 a 7, añadiendo gota a gota hidróxido de sodio 0,1 N. Transferir la solución cuantitativamente a un matraz aforado de 1.000 ml y enrasar con agua desionizada.
El pH después del ajuste deberá estar próximo a 6,5, lo que permite cualquier ligera variación durante el almacenamiento.
Usar agua desionizada.
1.4 Procedimiento.
1.4.1 Emulsionabilidad.
En una probeta graduada de 100 ml poner agua patrón a 30 ± 1 °C (1.5.3) hasta la marca de los 95 ml. Verter el concentrado emulsionable (1.5.4) poco a poco (5 ml de una probeta graduada) sobre la superficie del agua, colocar el tapón e invertir la probeta una vez (1.5.5).
Después de treinta segundos observar si la mezcla se ha emulsionado espontáneamente, dando 100 ml de una emulsión que aparece al examen visual como uniforme. Anotar si se produce espuma.
1.4.2 Estabilidad de la emulsión en reposo.
Invertir la probeta diez veces (1.5.5) y dejar la probeta y su contenido en reposo en el baño de agua a 30° ± 1 °C (1.5.3) durante veinticuatro horas.
Anotar el volumen (1.5.6), si existe, de aceite libre (1.5.7), espuma y crema formados tanto en la parte superior como en el fondo de la probeta, a los treinta minutos y a las dos y veinticuatro horas.
1.4.3 Reemulsión tras un reposo de veinticuatro horas.
Al cabo de las veinticuatro horas, invertir la probeta diez veces (1.5.5). Dejar en reposo durante treinta segundos y observar si el aceite, espuma, crema o materias sólidas encontradas tras el reposo de veinticuatro horas se han reemulsionado, dando 100 ml de una emulsión que aparece al examen visual (1.5.6) como uniforme.
1.4.4 Estabilidad final de la emulsión.
Dejar en reposo la probeta durante un plazo adicional de treinta minutos.
Anotar el volumen, si existe, de aceite libre, espuma, crema o materias sólidas presentes al cabo del período de treinta minutos.
1.5 Observaciones.
1.5.1 La probeta debe estar limpia y libre de grasa.‒Limpiar la probeta con una solución acuosa que contenga 5 por 100 de Ácido fluorhídrico, 30 por 100 de ácido nítrico y 2 por 100 de Teepol. Agitar durante veinte segundos. Enjuagar con agua destilada y escurrir.
Precaución.‒La mezcla anterior es corrosiva y no debe ponerse en contacto con la piel. No dejar la mezcla en la probeta durante más de treinta segundos.
1.5.2 La probeta deberá estar sujeta de tal forma que no esté en contacto con el recipiente del baño. La armadura del motor no deberá sujetarse a las paredes del baño.
1.5.3 A menos que se especifique otra cosa.
1.5.4 El concentrado emulsionable debe estar a la misma temperatura que el agua patrón que se va a usar para la dilución (30° ± 1 °C si no se indica otra cosa).
1.5.5 La expresión «invertir la probeta», como se usa más arriba, indica que la probeta tapada se inclina a mano 180 grados y luego se vuelve a su posición inicial, completando la operación en unos dos segundos, aproximadamente.
1.5.6 Para iluminar la probeta debe usarse una lámpara ajustable provista de una bombilla perlada de 60 watios. La posición y el ángulo de la luz deben ajustarse para conseguir la visión óptima de los límites de las fases. Con frecuencia es más fácil de ver esto con luz reflejada que con luz incidente.
1.5.7 Si inicialmente se encuentra dificultad para distinguir entre aceite y crema, puede usarse un colorante soluble en la fase oleosa, pero los ensayos finales deben hacerse sin la adición de colorantes. Se ha comprobado que los colorantes que dan una coloración azul oscura en los hidrocarburos aromáticos son los más adecuados para este propósito. El colorante (0,1 por 100) debe añadirse al concentrado emulsionable antes de hacer el ensayo. Si existe aceite, el colorante lo coloreará de azul oscuro; si se ha producido crema, el colorante dará una capa azul pálido; si no se ha producido crema o muy poca, no se originará ninguna banda de color definido.
1.6 Referencia.
1. CIPAC. Ed. 1970. MT-36 y MT-18.
2. ESTABILIDAD DE UNA EMULSIÓN
(Dilución de aplicación)
2.1 Principio.
Como en 1.1 de los métodos oficiales de análisis físicos.
El procedimiento descrito en este método es de aplicación a los concentrados emulsionables, que al ser diluidos a sus dosis de aplicación den lugar a emulsiones del tipo de aceite en agua.
2.2 Material y aparatos.
2.2.1 Vasos de precipitados de 250 ml con un diámetro interior de 6 cm.
2.2.2 Buretas de 50 ml, graduadas en 0,1 ml.
2.2.3 Probetas de 100 ml, graduadas en 1,0 ml.
2.2.4 Baño de agua (como 1.2.2).
2.3 Reactivos.
2.3.1 Agua patrón (como 1.3.1).
2.4 Procedimiento.
A 70 ml de agua patrón, colocados en un vaso de precipitados a la temperatura especificada (2.5.2), añadir, por medio de una bureta, el producto a ensayar a velocidad de unos 5 ml cada doce segundos. Durante la adición, agitar con una varilla de vidrio (a la velocidad de unas tres vueltas por segundo) y dirigir constantemente el flujo del concentrado hacia el centro y no contra la pared del vaso. Añadir la cantidad suficiente de concentrado para que cuando a continuación se lleve el volumen total a 100 ml se obtenga la dilución recomendada para la aplicación por el suministrador.
Llevar el volumen a 100 ml, añadiendo agua patrón con otra bureta. Transferir inmediatamente la emulsión diluida a una probeta graduada limpia y seca y mantener a ésta, así como el contenido, en reposo absoluto a la temperatura especificada ± 1 °C (2.5.2) durante una hora. Anotar cualquier cambio que se observe en la emulsión diluida y el volumen de cualquier materia que se hubiera separado.
Repetir la operación por completo utilizando agua destilada en lugar de agua patrón.
2.5 Observaciones.
2.5.1 A menos que se especifique otra cosa.
2.5.2 Si no se especifica otra cosa la temperatura debe ser 30° ± 1 °C.
2.6 Referencia.
1. CIPAC. Ed. 1970. MT-20.
3. SUSPENSIBILIDAD DE POLVOS MOJABLES
3.1 Principio.
La susceptibilidad se define como la cantidad de materia activa suspendida, después de un tiempo dado en una columna de líquido de alguna determinada, expresada como porcentaje de calidad de materia activa en la suspensión original.
Preparar una suspensión de concentración conocida en agua patrón o agua destilada, colocar dentro de una probeta a temperatura constante y dejar reposar durante cierto tiempo. Extraer 9/10 partes superiores y determinar el contenido de materia activa en la 1/10 parte restante, calculando después el contenido de las 9/10 partes superiores.
El método es aplicable para suspensiones que contengan hasta el 1 por 100 de materia activa.
3.2 Material y aparatos.
3.2.1 Probeta de 250 ml de tapón esmerilado (fig. 3.1).‒La distancia entre las graduaciones 0 y 250 ml deberá estar entre 20 y 21,5 cm y entre la señal de 250 ml y el tapón deberá existir una distancia de 4 a 6 cm.
3.2.2 Tubo de vidrio de succión de 40 cm de longitud y 5 mm de diámetro interior, finalizando por un extremo con una anchura de 2 a 3 mm; el extremo del tubo se conecta al vacío.
3.2.3 Vasos de precipitados de 250 ml.
3.2.4 Cronómetro.
3.2.5 Baño de agua a 30º ± 1 ºC.
3.3 Reactivos.
Como en 1.3.1.
3.4 Procedimiento.
3.4.1 Preparación de la suspensión.
Pesar suficiente cantidad de muestra para hacer 250 ml de suspensión en agua, a una concentración recomendada en las normas de uso sugeridas para el producto. Cuando se recomiendan distintas concentraciones se debe realizar la determinación de suspensibilidad a las concentraciones máxima y mínima. La muestra se dispersa con o sin papilla, de acuerdo con las normas de uso que acompañan al producto. Si no se dice lo contrario, se usará el método 3.4.1.2.
3.4.1.1 Con papilla.‒Colocar la muestra pesada en un vaso, añadir una pequeña cantidad de agua patrón (alrededor de 5 ml) y mezclar durante dos minutos con una varilla de vidrio con tope de goma, hasta producir una pasta uniforme. Añadir agua patrón (50 ml a 30° ± 1 ºC), gota a gota, mientras se remueve la mezcla y dejar la suspensión en reposo durante trece minutos, en un baño de agua a la misma temperatura.
3.4.1.2 Sin papilla.‒Añadir, lentamente, la muestra pesada al vaso que contiene el agua patrón 150 ml a 30° ± 1 °C). Agitar manualmente con movimiento circular a razón de 2 veces por segundo, durante dos minutos. Dejar reposar la suspensión durante trece minutos en un baño de agua a la misma temperatura.
3.4.2 Determinación de la sedimentación.‒Transferir la suspensión preparada en 3.4.1, cuantitativamente, a una probeta que haya sido calentada previamente a 30 °C; enrasar a 250 ml con agua patrón a 30º ± 1 ºC y poner el tapón. Manualmente, invertir la probeta 30 veces en un minuto.
Colocar la probeta en posición vertical y libre de toda vibración en un baño de agua, sin que le dé directamente la luz del sol. Después de treinta minutos (salvo que se especifique lo contrario) separar 225 ml (9/10 partes) del contenido en diez a quince segundos, usando el tubo de vidrio de absorción conectado a vacío, teniendo cuidado de no agitar o remover el sedimento de la probeta. Asegurarse de que el final del tubo de succión vaya siempre unos pocos milímetros por debajo de la superficie del líquido (3.6.2).
3.4.3 Determinación de la materia activa.‒Determinar el contenido de materia activa en la muestra original y en los 25 ml restantes de la probeta, por el método dado en los métodos de análisis prescritos para cada pesticida.
3.5 Cálculos.

Donde:
C = peso, en g, de la materia activa en la muestra tomada

a = tanto por ciento de materia activa determinada en la muestra antes o después del almacenamiento acelerado, según sea lo apropiado.
b = peso, en g, de la muestra.
Q = peso, en g, de la materia activa en los 25 ml restantes en la probeta.
3.6 Observaciones.
3.6.1 A menos que se especifique otra cosa, la prueba deberá realizarse en agua patrón.
3.6.2 El tubo de succión deberá ser conectado a través de una llave de dos vías a un frasco de seguridad. La llave deberá cerrarse suavemente, cuando las 9/10 partes de la suspensión hayan sido separadas, para evitar remover en la 1/10 parte restante.
3.7 Referencia.
1. CIPAC. Ed. 1970. MT-15.
4. DETERMINACIÓN DEL PUNTO DE FUSIÓN
4.1 Principio.
La muestra se calienta en tubo capilar a velocidad controlada en un baño líquido con agitador y se observan las temperaturas de formación del menisco y de total licuefacción de la muestra.
Punto de menisco es la temperatura a la cual tiene lugar la licuefacción de la sustancia, lo cual viene indicado por la formación de un menisco definido.
Punto de licuefacción es la temperatura a la cual la sustancia está completamente fundida, lo que se observa por la desaparición de la fase sólida.
Intervalo de fusión es el intervalo entre la temperatura a la cual la sustancia comienza a contraerse o a formar gotas en la pared del tubo capilar y la temperatura a la que la sustancia está completamente fundida, que es el punto de licuefacción.
4.2 Material y aparatos.
4.2.1 Aparato de punto de fusión (fig. 4.1).
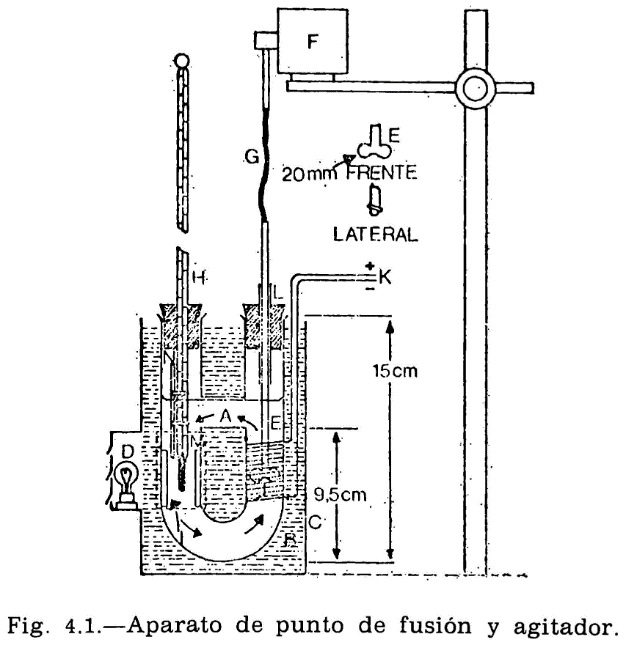
4.2.1.1 Descripción del aparato para determinación del punto de fusión:
A) Tubo de vidrio en U, conteniendo fluido de silicona.
B) Material rígido para fijación.
C) Caja de aluminio o latón.
D) Lámpara de 6 voltios, 0,3 A. Abertura de 4 x 1,5 cm en un lado de la caja metálica para transmitir luz a la muestra.
E) Agitador de vidrio de eje de 6 mm de ∅.
F) Motor de unas 375 r.p.m.
G) Goma flexible.
H) Termómetro contrastado de 0º a 300° graduado en divisiones de 1 ºC (4.6.2).
I) Camisa de vidrio de 3,8 cm de longitud y 0,3 mm de separación a las paredes, apoyada en los topes salientes.
J) Resistencia de calentamiento arrollada sobre papel de amianto.
K) Alimentación eléctrica desde una fuente de voltaje variable.
L) Camisa de tubo de vidrio para el eje del agitador.
M) Ventana de observación en la caja metálica.
N) Tubo capilar conteniendo la muestra y ajustando al termómetro por medio de un anillo de goma. Deberá estar seco, de pared delgada, de vidrio borosilicato, cerrado por un extremo y de diámetro interior de 1 mm aproximadamente, de 0,1 a 0,5 mm de espesor de pared y de una longitud suficiente para que el extremo abierto quede por encima de la superficie del líquido en el tubo de calentamiento. Como mínimo, deberá tener 12 cm de longitud. Los capilares se guardarán cerrados por ambos extremos, cortándolos tan solo cuando se precisen.
El aparato consiste en un tubo de vidrio en U con tubo de conexión superior entre las dos ramas, de forma que un líquido, calentado por una resistencia externa y propulsado y mezclado por un agitador situado en una de las ramas, circula alrededor del tubo con la muestra y del termómetro colocados en la segunda rama. La muestra colocada en un tubo capilar se ajusta al termómetro de modo que el bulbo y el tubo estén situados juntos en el baño en una posición fija para su observación. Una lámpara ilumina la muestra durante la determinación.
Los circuitos eléctricos para controlar la velocidad de calentamiento y la circulación en el baño líquido se describen en la figura 4.2 y deben ser convenientemente montados en una caja metálica. Fijando el aparato del punto de fusión a la superficie superior de la caja metálica se puede trasladar como una sola unidad.
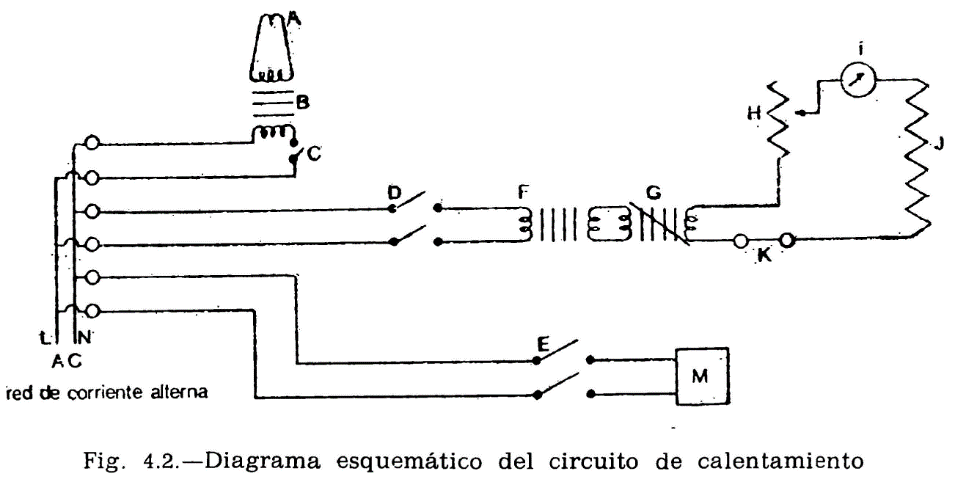
A) Lámpara de 6 voltios, 0,3 A.
B) Transformador de 6,3 voltios, 0,5 A.
C) Interruptor monofásico.
D) y E) Interruptores bifásicos.
F) Transformador de 48 voltios, 2 A de salida.
G) Transformador de voltaje de salida variable. Si está calibrado en su voltaje de salida, el transformador F, los interruptores D y C y el amperímetro I pueden omitirse.
H) Resistencia variable de 8 a 10 ohms.
I) Amperímetro de 2 A de corriente alterna.
J) Resistencia de calentamiento.
K) Fusible de 2 amperios.
M) Motor de unas 375 r.p.m.
4.2.1.2 Construcción, del aparato para la determinación del punto de fusión:
Construir el tubo en U, de 2,5 cm de diámetro, de vidrio, borosilicato o similar. Poner una capa de amianto, en forma de papel o cinta, alrededor de la parte inferior de una de las ramas y enrollar sobre ella una resistencia de 7 ohms por metro, en una longitud de 4 cm sobre el tubo y con 4 espirales por cm. La resistencia total debe ser de unos 25 ohms.
Fijar firmemente el tubo en U dentro de un recipiente metálico de aproximadamente 15 x 10 x 5 cm, usando un material rígido aislante tal como fibra de amianto o lana de vidrio. Adaptar a la caja un soporte lateral conteniendo una pequeña lámpara eléctrica, de forma que la muestra se ilumine a través de una abertura en el recipiente principal y las observaciones puedan realizarse por medio de una segunda abertura en la parte frontal de la caja.
Montar el motor de agitación sobre la rama del tubo U provista de la resistencia de calentamiento mediante un soporte adecuado. Colocar el agitador de vidrio en el tubo en U y acoplarlo por medio de un trozo de goma al eje del motor. El vástago de vidrio giratorio se introduce por medio de una guía de vidrio a través del corcho de la rama del tubo en U. El tubo capilar y el bulbo del termómetro se colocan en el centro de la parte visible de la segunda rama del tubo, por medio de un corcho. En esta posición, la muestra queda totalmente iluminada por la lámpara y puede ser observada claramente a través de la ventana de la parte frontal.
4.3 Reactivos.
4.3.1 Fluido de silicona (4.6.1).
4.4 Procedimiento.
4.4.1 Calibrado del aparato.
Llenar el tubo en U con fluido de silicona de un tipo adecuado (4.6.1) hasta el nivel indicado en la figura 4.1. Con el agitador y el termómetro en posición poner en marcha el agitador, haciendo pasar una corriente de 0,1 A, a través de la resistencia. Leer la temperatura del baño a intervalos fijos. Después, comenzando por 0,1 A para la corriente de calentamiento y observando el ascenso de temperatura hasta que se alcance la máxima, repitiendo el incremento de 0,1 A, se pueden obtener una serie de curvas de calentamiento, figura 4.3.
El examen de estas curvas de calentamiento indicará para cada intervalo de temperaturas la intensidad de corriente necesaria para conseguir una velocidad de calentamiento de 2 ºC por minuto.
4.4.2 Determinación del punto de fusión.
Preparar la muestra pulverizándola en un mortero, secarla en capa delgada a la temperatura y tiempo indicados y volver a moler si es necesario. Por otra parte, cuando la temperatura de desecación no se especifica, secarla a una temperatura bastante inferior a su punto de fusión, bajo presión reducida o bien sobre un desecante apropiado. Guardar la muestra en un desecador.
Pasar una porción a uno de los tubos capilares, agitarlo de forma que pase el polvo hacia la extremidad cerrada y comprimirlo ligeramente golpeando el tubo sobre una superficie dura, para que forme una columna de 2 a 4 mm de altura (4.6.3.).
Sujetar el capilar al termómetro con una anilla de goma y, si fuera necesario, otro tubo similar conteniendo la muestra patrón, de forma que la muestra quede situada en la mitad del bulbo. Colocarlos en la rama izquierda del tubo en U, de tal forma que el nivel del líquido llegue a la marca de inmersión. Conectar a la red del motor del agitador, el circuito de calentamiento y la lámpara para iluminar la muestra. Hallar el punto de fusión aproximado y seleccionar el grado apropiado de calentamiento a partir de las curvas calibradas. Ajustar el calentamiento, a razón de 3 a 5 ºC por minuto, hasta alcanzar unos 5 ºC por debajo del punto de fusión y entonces disminuir la corriente eléctrica para lograr la subida específica de temperatura. Si no está especificada la velocidad de calentamiento, adoptar la de 2 ºC por minuto.
Para la observación del punto de fusión es muy conveniente utilizar una lupa manual o fija.
4.5 Cálculo.
La temperatura de la parte emergente del termómetro se determina colocando el bulbo de un segundo termómetro en contacto con la parte que emerge y en un lugar de ésta situado aproximadamente en el punto medio de la columna de mercurio que sobresale del líquido.
La corrección viene dada por la siguiente ecuación:
Tc = 0,00016 n (ts - td)
Siendo:
tc = corrección para ser aplicada a la temperatura observada del punto de fusión.
ts = temperatura media de la columna emergente durante la calibración.
td = temperatura media de la columna emergente observada en el punto de fusión.
n = longitud de la columna descubierta, medida en grados centígrados.
La temperatura corregida se toma como el punto de fusión de la sustancia.
A menos que se indique otra cosa, el punto de menisco se toma como el punto de fusión de la muestra. Cuando el punto de fusión en la especificación o método de análisis viene dado como un intervalo, el punto de fusión de la sustancia deberá estar comprendido en dicho intervalo.
4.6 Observaciones.
4.6.1 Para el intervalo de temperaturas de 20° a 200 °C es adecuado un fluido de silicona de 20 centipoises. Para temperaturas hasta 300° debe utilizarse un fluido de silicona de 50 centipoises.
4.6.2 Para mayor precisión en las lecturas se usarán termómetros contrastados graduados en divisiones de 0,2 °C y de escalas adecuadas.
4.6.3 Cuando se determinan puntos de licuefacción, esta longitud no debe sobrepasarse, ya que pueden cometerse errores superiores a 0,8 °C.
4.7 Referencia.
1. CIPAC Ed. 1970. MT-2.
5. DETERMINACIÓN DEL PUNTO DE FUSIÓN DEL ÁCIDO 2,4 DICLORO FENOXIACETICO PROCEDENTE DE ÉSTER
5.1 Principio.
Por saponificación del éster se aísla el ácido y se determina su punto de fusión como en 4.1.
5.2 Material y aparatos.
5.2.1 Matraz Erlenmeyer de 250 ml con boca esmerilada.
5.2.2 Refrigerante de reflujo, cuyo extremo inferior esté cortado recto y acabe en la parte esmerilada de la boca del matraz para que el reflujo lave las paredes del mismo.
5.2.3 Embudos de decantación de 250 ml.
5.2.4 Cápsula de evaporación.
5.2.5 Probeta graduada de 10 ml.
5.2.6 Estufa regulable a 105 °C.
5.2.7 Baño de agua.
5.2.8 Aparato para la determinación del punto de fusión. Como 4.2.
5.2.9 Placa de calefacción con agitador magnético.
5.2.10 Matraz aforado de 250 ml.
5.3 Reactivos.
5.3.1 Ácido clorhídrico concentrado.
5.3.2 Éter etílico.
5.3.3 Etanol del 95 por 100.
5.3.4 Hidróxido de litio 1 N.
5.4 Procedimiento.
5.4.1 Calibrado del aparato. Como en 4.4.1.
5.4.2 Determinación del punto de fusión.
Pesar la cantidad de muestra necesaria para obtener 0,5 g de ácido, 2,4 D en un matraz Erlenmeyer de 250 ml. Adicionar 5 ml de etanol y 12,5 ml de solución de hidróxido de litio 1 N. Colocar el refrigerante y calentar con agitación durante una hora a reflujo intenso. Enfriar y trasvasar con agua destilada a un embudo de decantación de 250 ml. El volumen final será de unos 60 ml.
Extraer con 15 ml de éter etílico, dejando un tiempo de decantación de treinta minutos (hasta obtener una separación total de las dos fases) y transvasar la fase acuosa a un matraz aforado de 250 ml. Lavar la fase etérea con (2 x 5 ml) de agua, dejando un tiempo de decantación de quince minutos después de cada lavado.
Trasvasar las soluciones de lavado al matraz aforado y enrasar con agua destilada y agitar. Desechar la fase etérea. Tomar con pipeta 100 ml y llevar a un embudo de decantación de 250 ml. Añadir 1,5 ml de ácido clorhídrico y extraer tres veces con 12,5 ml de éter etílico cada vez, dejando en cada decantación un tiempo de reposo de quince minutos.
Pasar la totalidad de los extractos etéreos a la cápsula de evaporación y evaporar el éter en baño de agua. Desecar el residuo en estufa a 105 °C, durante cuatro horas. Desprender, cuanto sea posible, el ácido de la cápsula, mediante una espátula y mezclar cuidadosamente el ácido pulverizado. Continuar como en 4.4.2.
5.5 Cálculo.
Como en 4.5.
5.6 Referencia.
1. CIPAC. Ed. 1970. 1.3/5/M/1.5.
6. DETERMINACIÓN DEL PUNTO DE FUSIÓN DEL ÁCIDO 2,4 DICLORO FENOXIACÉTICO PROCEDENTE DE SAL AMINA
6.1 Principio.
Por acidificación de la sal, se aísla el ácido y se determina su punto de fusión como en 4.1.
6.2 Material y aparatos.
Como en 5.2.1, 5.2.3, 5.2.4, 5.2.5, 5.2.6, 5.2.7, 5.2.8.
6.3 Reactivos.
Como en 5.3.1, 5.3.2.
6.4 Procedimiento.
6.4.1 Calibrado del aparato como en 4.4.1.
6.4.2 Determinación del punto de fusión.
Pesar la cantidad de muestra necesaria para obtener 0,5 g del ácido 2,4 D en un matraz Erlenmeyer de 250 ml. Diluirlo con agua destilada hasta un volumen de unos 80 ml y traspasarla a un embudo de decantación de 250 ml, empleando un volumen de unos 20 ml de agua destilada para hacer el traspaso cuantitativo. Añadir 1,5 ml de ácido clorhídrico y extraer tres veces con 12,5 ml de éter etílico cada vez, dejando en cada decantación un tiempo de reposo de quince minutos.
El conjunto de los extractos etéreos se lava tres veces con 10 ml de agua destilada cada vez, dejando el tiempo de decantación necesario para tener una separación total de las fases. Al final de estos lavados, comprobar que el pH de la última fase acuosa separada no ha variado. Trasvasar la fase etérea a un matraz Erlenmeyer de 250 ml. Pasar las aguas de lavado al embudo de decantación y efectuar una extracción con 30 ml de éter etílico, dejando un tiempo de decantación de una hora.
Desechar la fase acuosa, lavar dos veces la fase etérea, con 25 ml de agua destilada cada vez, dejando un tiempo de decantación de treinta minutos y comprobando que su pH es, aproximadamente 7. Reunir esta fase etérea con la anterior, situada en el matraz Erlenmeyer.
Pasar la totalidad de los extractos etéreos a la cápsula de evaporación y evaporar el éter en baño de agua. Desecar el residuo en estufa a 105 °C durante cuatro horas. Desprender, cuanto sea posible, el ácido de la cápsula mediante una espátula y mezclar cuidadosamente el ácido pulverizado. Continuar como en 4.4.2.
6.5 Cálculo.
Como en 4.5.
6.6 Referencia.
1. CIPAC. Edición 1970. 1.4/13/M/1.5.
7(a). HUMEDAD
(Método de Karl-Fischer)
7(a).1 Principio.
Dispensar la muestra en metanol y valorar con reactivo de Karl-Fischer de equivalente en agua conocido.
7(a).2 Material y aparatos.
7(a).2.1 Bureta automática de 25 ml provista de un sistema de desecación de aire [fig. 7 (a).1].
7(a).2.2 Vaso de reacción de unos 60 ml de capacidad, con dos electrodos de platino, un tubo de entrada de nitrógeno, un tapón que permite el paso de la bureta, un tubo de ventilación protegido por un desecante y una boca de carga con tapón esmerilado.
La muestra a valorar se introduce a través de la boca lateral. La agitación se mantiene por una corriente de nitrógeno seco a través de la solución o magnéticamente. Si se usa nitrógeno, emplear tres frascos de secado, uno con ácido sulfúrico y dos con gel de sílice activado recientemente (en este orden).
7(a).2.3 Pila de 1,5 a 2 voltios, conectada a través de una resistencia variable de 2.000 ohmios. La resistencia se ajusta de manera que una corriente inicial de no más de 20 milivoltios pase a través de los electrodos de platino, conectados en serie con un microamperímetro, cuando hay un exceso de 0,2 ml de reactivo de Karl-Fischer. Después de cada adición del reactivo de Karl-Fischer la aguja del galvanómetro se mueve, pero rápidamente vuelve a su posición inicial. En el punto final se obtiene una desviación que tarda más tiempo en volver a su posición inicial.
7(a).2.4 Galvanómetro con una escala de desviación no mayor que 100 microamperios.
7(a).3 Reactivos.
7(a).3.1 Metanol que no contenga más del 0,03 por 100 de agua (p/p).
7(a).3.2 Iodo resublimado.
7(a).3.3 Nitrógeno seco.
7(a).3.4 Piridina que no contenga más del 0,1 por 100 de agua (p/p)
7(a).3.5 Anhídrido sulfuroso líquido, de calidad para refrigeración.
7(a).3.6 Reactivo de Karl-Fischer.
7(a).3.6.1 Preparación.‒Disolver 63 g de iodo en 100 ml de piridina seca, enfriar con hielo y hacer pasar anhídrido sulfuroso a través de la mezcla, hasta que la solución aumente su peso en 32,3 g. Evitar la absorción de la humedad atmosférica. Añadir metanol suficiente para obtener 500 ml de solución y dejar en reposo durante veinticuatro horas
7(a).3.6.2 Normalización.‒Añadir alrededor de 20 ml de metanol al vaso y reactivo hasta el punto de equilibrio, sin anotar el volumen. Introducir una cantidad exacta de agua, pesando la proporción adecuada de tartrato sódico dihidratado en polvo que contenga 15,61 a 15,71 por 100 de agua (300-500 mg) y valorar de nuevo con el reactivo de Karl-Fischer. Calcular el equivalente en agua del reactivo en mg de agua por ml. Teniendo en cuenta que el reactivo de Karl-Fischer degrada rápidamente deberá ser normalizado inmediatamente antes de su uso, o diariamente. Cuando la preparación es reciente 1 ml equivale a 5 mg de agua aproximadamente.
7(a).4 Procedimiento.
Añadir alrededor de 20 ml de metanol al vaso de valoración y valorar hasta el punto de equilibrio con el reactivo de Karl-Fischer. Transferir rápidamente una cantidad adecuada de muestra exactamente pesada (P) al vaso de valoración. Agitar durante un minuto y valorar de nuevo con el reactivo de Karl-Fischer (V) de equivalente en agua conocido.
7(a).5 Cálculos.

Siendo:
V = volumen, en ml, de reactivo de Karl-Fischer gastados en la valoración.
P = peso, en g, de la muestra.
C = equivalente en agua, en mg/ml, del reactivo de Karl- Fischer.
7(a).6 Referencias.
1. Collaborative International Pesticides Analytical Council Limited (CIPAC). Ed. 1970. MT/30.1.
7(b). HUMEDAD
(Método de Dean-Stark)
7(b).1 Principio.
El agua de la muestra se determina por destilación azeotrópica con tolueno, xileno o solvente nafta.
Este método sólo es recomendable para productos cuyos contenidos de humedad sean superiores al 2 por 100.
7(b).2 Material y aparatos.
7(b).2.1 Aparato de Dean Stark (fig. 7(b).I).
7(b).2.1.1 Refrigerante recto.
7(b).2.1.2 Colector de 2 ml con divisiones de 0,05.
7(b).2.1.3 Matraz de 500 ml.
7(b).2.2 Probeta graduada de 200 ml.
7(b).3 Reactivos.
7(b).3.1 Tolueno.
7(b).3.2 Xileno.
7(b).3.3 Disolvente nafta. Punto de ebullición de 90 a 160 °C.
7 (b).4. Procedimiento
Pesar una cantidad de muestra que contenga de 0,5 a 1,5 ml de agua. Transferirla al matraz de 500 ml que contenga 100 ml de tolueno, a menos que se indique otro disolvente, y añadir 100 ml más de disolvente y porcelana porosa. Conectar el aparato y poner algodón en el extremo superior del refrigerante para prevenir la entrada de humedad atmosférica. Calentar el matraz hasta que el reflujo sea de 2 a 5 gotas por segundo y continuar la destilación hasta que el agua condensada no sea visible en ninguna parte excepto en el fondo del colector, y el volumen de agua recogida permanezca constante durante cinco minutos. Separar el agua adherida en el refrigerante, aumentando la velocidad de reflujo en unas cuantas gotas por segundo o bien lavando el refrigerante con tolueno. Dejar enfriar el aparato a temperatura ambiente y separar las gotas de agua adheridas a la parte superior del colector con un alambre fino. Leer el volumen de agua.
7(b).5 Cálculos.

Siendo:
V = volumen, en ml, de agua en el colector.
P = peso, en g, de la muestra.
Los resultados de las repeticiones no deben diferir en más de 0,025 ml.
7(b).6 Referencias.
1. Collaborative International Pesticides Analytical Council Limited (CIPAC). Ed. 1970. MT/30.2.
8. TAMAÑO DE GRÁNULOS
8.1 Principio.
Separación cuantitativa del producto en fracciones de partículas de diferentes tamaños empleando un tamiz o serie de tamices.
8.2 Material y aparatos.
8.2.1 Tamices para análisis de unos 20 cm de diámetro y de luz de malla de 850, 710, 500, 420, 355, 250 y 250 μ (tabla 8.I).
8.2.2 Tapa y colector para los tamices anteriores.
8.2.3 Pincel plano suave (2,5 cm) para la limpieza de las mallas más finas de los tamices.
8.2.4 Cepillo plano duro para la limpieza de las mallas más gruesas de los tamices.
8.2.5 Balanza con sensibilidad de 0,1 g y precisión de ± 0,05 g.
8.2.6 Agitador de tamices. La tabla del agitador para los tamices en la que se deposita la plataforma está inclinada respecto al eje un ángulo de 4,5 grados y su movimiento es como el de un disco inclinado que girará alrededor de su perímetro; la velocidad de la rotación es, aproximadamente, de 2,5 r.p.m. Este movimiento giratorio extiende el producto sobre las mallas de los tamices y al mismo tiempo la tabla vibra hacia arriba y abajo sobre una distancia de 4 mm, con una frecuencia de 300 vibraciones por minuto. Los movimientos de rotación y vibración son producidos per el mismo mecanismo, de ahí que sea suficiente medir la velocidad de rotación de la tabla inclinada para comprobar las características de cualquier vibrador. El período designado para el tamizado de gránulos es de cuarenta y cinco minutos, a 2,5 r.p.m., si no se cumple ajustarlo mediante la fórmula:

8.3 Procedimiento.
8.3.1 Preparación de la muestra.
Quitar las tapas de las cajas que contienen las muestras y secarlas en un desecador de vacío a 18 °C empleando pentóxido de fósforo como desecante hasta obtener peso constante.
8.3.2 Tamizado.‒Acoplar el juego de tamices en el orden correcto con el más fino en la parte más inferior y situado sobre el plato receptor.
Pesar la caja de muestra y sus contenidos. Vaciarla con cuidado sobre el tamiz más grueso (850 μ) habiendo limpiado con el cepillo cualquier residuo que hubiera en el tamiz. Pesar la caja que se ha vaciado y anotar el peso de la muestra transferida al tamiz. Poner la tapa al juego de tamices situándoles en la máquina agitadora. Agitar durante cuarenta y cinco minutos.
Quitar el juego de tamices de la máquina tras haber dejado reposar polvo durante unos dos minutos. Quitar con cuidado la tapa e invertir cada tamiz sobre una hoja de papel. Dar unos golpecitos suaves al tamiz y con cuidado cepillar su superficie superior. Volcar el tamiz y repetir echando fuera cualquier partícula que permaneciera. Añadir los cepillados al material obtenido en cada tamiz. Seguir este procedimiento con los tamices de 850 μ a 250 μ, inclusive, y anotar los pesos con aproximación de 0,1 g. Anotar también los pesos, con aproximación de 0,1 g del material recogido sobre el tamiz de 150 μ en el plato receptor.
8.3.3 Pérdida de polvo durante el tamizado.
Los pesos recogidos sobre los tamices, junto con los pesos del material recogido en el plato receptor, se sumarán juntos y se restarán del peso de la muestra original. Ello dará la pérdida durante el tamizado, que deberá ser menor del 0,25 por 100 del peso de la muestra. En caso contrario, repetir el ensayo.
Añadir el peso de la muestra perdida en el tamiz (inferior al 0,25 por 100 del peso de la muestra) a la fracción que pasó al tamiz de 150 μ.
8.4 Cálculos.
Expresar el peso de cada fracción de tamiz como un porcentaje del peso de la muestra, con aproximación del 0,1 por 100.
8.5 Referencias.
1 Collaborative International Pesticides Analytical Council Limited (CIPAC). Ed. 1970. MT/58.
TABLA 8.I
EQUIVALENCIAS ENTRE MEDIDAS DE MALLAS
Relación entre los tamices de la serie UNE 7050 y los de las principales series extranjeras
|
Número de referencia |
España | Alemania | USA | Inglaterra | Francia | URSS |
inter nacional |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
UNE 7050 Luz de malla mm |
DIN 1171 Luz de malla mm |
Número |
DIN 4148 Luz de malla mm |
ASTM Luz de malla mm |
Número |
TYLER Luz de malla mm |
Número |
ES 410 Luz de malla mm |
Número |
AFNOR XII-501 Luz de malla mm |
GOST Luz de malla mm |
1SO-TC-7 Luz de malla mm |
|
| 50 | 25 | ‒ | 25 | 25,4 | 25,4 | 1 | ‒ | ‒ | ‒ | ||||
| 51 | ‒ | ‒ | 22,2 | 22,43 | 22,23 | 7/8" | ‒ | ‒ | 22,4 | ||||
| 52 | 20 | ‒ | 20 | ‒ | ‒ | ‒ | ‒ | ‒ | ‒ | ||||
| 53 | ‒ | 18 | 19,1 | 18,85 | 19,05 | 3/4" | ‒ | ‒ | ‒ | ||||
| 54 | 16 | ‒ | 16 | 15,9 | ‒ | 15,88 | 5/8" | ‒ | ‒ | 16 | |||
| 55 | |||||||||||||
| 56 | 12,5 | ‒ | 12,5 | 12,7 | ‒ | 12,7 | 1/2" | ‒ | ‒ | ‒ | |||
| 57 | |||||||||||||
| 58 | 10 | 10 | 10 | ‒ | ‒ | ‒ | ‒ | ‒ | ‒ | ||||
| 59 | |||||||||||||
| 60 | 8 | 8 | 8 | 7,93 | 7,923 | 2,5 | ‒ | ‒ | ‒ | 8 | |||
| 61 | |||||||||||||
| 62 | 6,3 | 6 | 6,3 | 6,35 | 3 | ‒ | 6,35 | 3 | 6 | ‒ | ‒ | ||
| 63 | ‒ | ‒ | ‒ | 5,66 | 3,5 | ‒ | ‒ | ‒ | ‒ | ‒ | |||
| 64 | 5 | 5 | 5 | ‒ | ‒ | 5 | ‒ | 5 | |||||
| 65 | ‒ | ‒ | ‒ | 4,76 | 4 | ‒ | ‒ | ‒ | ‒ | ||||
| 66 | 4 | 4 | 4 | 4 | 5 | 3,962 | 5 | ‒ | 4 | ‒ | 4 | ||
| 67 | |||||||||||||
| 68 | 3,2 | 3 | 3,15 | 3,36 | 6 * | ‒ | 3,18 | 1/8" | 3,15 | ‒ | ‒ | ||
| 69 | ‒ | ‒ | ‒ | 2,83 | 7 | 2,794 | 7 | 2,812 | 6 | ‒ | ‒ | 2,8 | |
| 70 | 2,5 | 2,5 | 2,5 | 2,38 | 8 | 2,362 | 8 | 2,411 | 7 | 2,5 | 2,5 | ‒ | |
| 71 | |||||||||||||
| 72 | 2 | 2 | 3 | 2 | 2 | 10 | 1,981 | 9 | 2,057 | 8 | 2 | 2 | 2 |
| 73 | |||||||||||||
| 74 | 1,6 | ‒ | 1,6 | 1,68 | 12 * | 1,651 | 10 | 1,6 | 10 | 1,6 | 1,6 | ‒ | |
| 75 | 1,5 | 4 | ‒ | 1,41 | 14 | 1,397 | 12 | 1,405 | 12 | ‒ | ‒ | 1,4 | |
| 76 | 1,25 | ‒ | 1,25 | ‒ | ‒ | 1,25 | 1,25 | ‒ | |||||
| 77 | 1,2 | 5 | ‒ | 1,19 | 16 | 1,168 | 14 | 1,204 | 14 | ‒ | ‒ | ‒ | |
| 78 | 1 | 1 | 6 | 1 | 1 | 18 | 0,991 | 16 | 1,003 | 16 | 1 | 1 | 1 |
| 79 | |||||||||||||
| 80 | 0,80 | 0,80 | 0,84 | 20 * | 0,883 | 20 | 0,83 | 18 | 0,8 | 0,8 | ‒ | ||
| 81 | ‒ | 0,75 | 8 | ‒ | 0,71 | 25 | 0,701 | 24 | 0,699 | 22 | ‒ | 0,7 | 0,71 |
| 82 | 0,63 | 0,6 | 10 | 0,63 | 0,59 | 30 * | 0,589 | 28 | ‒ | 0,63 | 0,63 | ‒ | |
| 83 | ‒ | 0,54 | 11 | ‒ | ‒ | ‒ | ‒ | ‒ | ‒ | ‒ | |||
| 84 | 0,00 | 0,50 | 12 | 0,50 | 0,50 | 36 | 0,495 | 32 | 0,50 | 30 | 0,50 | 0,50 | 0,50 |
| 85 | 0,43 | 14 | ‒ | ‒ | ‒ | ‒ | ‒ | ‒ | ‒ | ||||
| 86 | 0,40 | 0,40 | 16 | 0,40 | 0,42 | 40 * | 0,417 | 35 | 0,422 | 36 | 0,4 | 0,4 | ‒ |
| 87 | ‒ | ‒ | 0,35 | 45 | 0,351 | 42 | 0,353 | 44 | ‒ | 0,355 | 0,355 | ||
| 88 | 0,32 | 0,3 | 20 | 0,315 | 0,297 | 50 * | 0,294 | 48 | ‒ | 0,315 | 0,315 | ‒ | |
| 89 | |||||||||||||
| 90 | 0,25 | 0,25 | 24 | 0,25 | 0,25 | 60 | 0,246 | 60 | 0,251 | 60 | 0,25 | 0,25 | 0,25 |
| 91 | |||||||||||||
| 92 | 0,20 | 0,20 | 30 | 0,20 | 0,210 | 70 * | 0,203 | 65 | 0,211 | 72 | 0,2 | 0,2 | ‒ |
| 93 | 0,177 | 80 | 0,175 | 80 | 0,178 | 85 | 0,18 | 0,18 | |||||
| 94 | 0,160 | 0,160 | ‒ | ‒ | 0,160 | 0,160 | ‒ | ||||||
| 95 | 0,150 | 40 | ‒ | 0,149 | 100 * | 0,147 | 100 | ‒ | ‒ | ‒ | ‒ | ||
| 96 | 0,125 | ‒ | 0,125 | 0,125 | 120 | 0,124 | 115 | 0,124 | 120 | 0,125 | 0,125 | 0,125 | |
| 97 | 0,120 | 50 | ‒ | ‒ | ‒ | ‒ | ‒ | ‒ | ‒ | ||||
| 98 | 0,100 | 0,100 | 60 | 0,100 | 0,105 | 140 * | 0,104 | 150 | 0,104 | 150 | 0,100 | 0,100 | ‒ |
| 99 | 0,090 | 70 | 0,090 | 0,088 | 170 | 0,088 | 170 | 0,089 | 170 | 0,090 | 0,090 | ||
| 100 | 0,080 | 0,080 | ‒ | ‒ | ‒ | ‒ | 0,080 | ‒ | |||||
| 101 | 0,075 | 80 | 0,071 | 0,074 | 200 * | 0,074 | 200 | 0,076 | 200 | ‒ | 0,071 | ‒ | |
| 102 | 0,063 | 0,060 | 100 | 0,060 | 0,062 | 230 | 0,062 | 230 | ‒ | ‒ | 0,063 | 0,063 | |
| 103 | 0,056 | ‒ | ‒ | ‒ | ‒ | 0,056 | ‒ | ||||||
| 104 | 0,050 | 0,050 | 110 | 0,050 | 0,053 | 270 * | 0,068 | 270 | 0,063 | 300 | 0,050 | 0,050 | ‒ |
| 105 | ‒ | 0,045 | 0,045 | 0,044 | 325 | 0,043 | 325 | 0,044 | 350 | ‒ | 0,045 | 0,045 | |
| 106 | 0,040 | 0,040 | 130 | 0,040 | ‒ | ‒ | 0,040 | ‒ | |||||
| 107 | ‒ | 0,037 | 150 | ‒ | 0,037 | 400 | 0,037 | 400 | ‒ | ‒ | ‒ | ‒ | |
* Tamices AFA en la serie ASTM.
Luces de malla especiales en telas de nylon: 0,035, 0,030, 0,028, 0,025, 0,023, 0,020, 0,012 mm.
9. FINURA EN POLVOS
(Por vía seca)
9.1 Principio.
Separación cuantitativa de un polvo en fracciones de partículas de diferentes tamaños empleando un tamiz o series de tamices.
9.2 Material y aparatos.
9.2.1 Tamices para análisis, de unos 20 cm de diámetro, excepto si se especifica otra cosa, y tamaño de malla apropiado (tabla 8.1).
9.2.2 Colectores y tapas que ajusten.
9.2.3 Pincel plano y suave de 2,5 cm.
9.2.4 Vidrios de reloj tarados.
9.3 Procedimiento.
9.3.1 Preparación de la muestra.‒Si se apreciara humedad en la muestra y si el desecado no está expresamente excluido en el método prescrito para el producto, desecar una cantidad adecuada a 100 °C hasta peso constante (o a una temperatura inferior si el método lo requiere, o si fuera necesario debido a las propiedades físicas de alguno de los componentes). Antes de pesar la muestra para el análisis, dejar que el producto se equilibre con la humedad ambiente, a no ser que el polvo sea altamente higroscópico, en cuyo caso debe enfriarse en un desecador y realizar el subsiguiente tamizado exponiéndolo lo menos posible a la atmósfera.
9.3.2 Extracción preliminar del polvo fino.‒Escoger los tamices del tamaño de malla requerido por la especificación (9.5.1), ajustar el tamiz más fino (9.5.2) sobre el recipiente colector y poner dentro del tamiz una parte pesada de la muestra preparada. Ajustar la tapa y agitar el conjunto con un movimiento oscilatorio, golpeando alternativamente los lados izquierdo y derecho del fondo del recipiente sobre una superficie de madera y haciéndolo girar al mismo tiempo con las manos. A intervalos examinar el tamiz, dejando que el polvo se sedimente durante unos segundos antes de quitar la tapa y emplear el pincel para quitar cualquier material que obstruya las mallas (9.5.3) hasta que el producto no tenga polvo fino. Desechar el producto que ha pasado por el tamiz.
9.3.3 Tamizado.‒Montar en columna el juego de tamices requeridos, en orden decreciente según las medidas de las mallas, siendo el superior el de paso más grueso y poner en el fondo el recipiente colector. Transferir el residuo procedente del tamizado previo al tamiz superior, ajustar la tapa y efectuar el tamizado como en 9.3.2. Continuar hasta que la cantidad de residuo del tamiz superior sea constante. No se debe intentar machacar las partículas duras, pero deben desmenuzarse con el pincel los agregados blandos.
Quitar el tamiz superior (el más grueso), ajustarle la tapa y un segundo recipiente colector, reanudar el tamizado con este tamiz y a intervalos de dos minutos examinar cualquier producto que haya pasado el tamiz y transferirlo al siguiente de la serie. Continuar con el mismo tamiz hasta que no pase más material (en el tiempo de dos minutos) o sólo pase una cantidad insignificante (9.5.4).
Repetir con cada uno de los sucesivos tamices la técnica descrita para el tamiz más grueso, desechando lo que pase de nuevo por el de malla más fina. Transferir cada residuo a vidrios de reloj tarados y pesar.
9.4 Cálculos.
Para ensayos con un solo tamiz, expresar el peso del residuo en tanto por ciento respecto al peso de la muestra y anotar el resultado como porcentaje de retención en el tamiz de ensayo establecido.
Cuando haya de especificar en más de un tamiz, añadir al porcentaje del residuo de cada tamiz, los porcentajes de residuos, igualmente calculados, en todos los tamices superiores de la serie. Anotar la suma total como el porcentaje de retención en el tamiz de ensayo establecido.
9.5 Observaciones.
9.5.1 Las telas metálicas de los tamices de mallas finas están hechas a base de bronce fosforoso. Este material no es adecuado para tamizar organomercuriales y a este fin se necesitan telas metálicas de acero inoxidable.
9.5.2 Cuando se requiera sólo una medida de malla, emplear el tamiz específico como se indica en este párrafo y completar el tamizado como se describe para cada tamiz por separado (9.3.3).
9.5.3 Los tiempos relativos que se emplean en dar pequeños golpes y cepillar y el tiempo total empleado variará de un tipo de polvo a otro y la técnica más efectiva para un determinado polvo se consigue sólo a base da adquirir experiencia con dicho producto.
9.5.4 En muchos casos continuarán pasando indefinidamente cantidades mínimas del producto y se necesita tener experiencia con el producto para establecer el punto final de tamizado. En algunos procedimientos de tamizado se considera concluido cuando no pasa más de 0,2 por 100 en peso de la muestra en dos minutos.
9.6 Referencias.
1. Collaborative International Pesticides Analytical Council Limited (CIPAC). Ed. 1970. MT/59.1.
10. FINURA DE POLVOS
10.1 Principio.
Separación cuantitativa de un polvo en fracciones de partículas de diferentes tamaños empleando los tamices apropiados y arrastrando el producto por una corriente de agua.
10.2 Material y aparatos.
10.2.1 Vaso de 250 ml.
10.2.2 Varilla de vidrio con protector de goma.
10.2.3 Tamices de unos 10 cm de diámetro y de malla adecuada. Tabla 8.I.
10.3 Reactivos.
10.3.1 Mojante no iónico.
10.4 Procedimiento.
10.4.1 Mojado del polvo.‒En un vaso de 250 ml pesar exactamente una cantidad adecuada (unos 20 g) de la muestra y añadirle suficiente volumen de agua, mientras se remueve con una varilla de vidrio cubierta de un protector de goma, hasta formar una pasta clara. No se debe intentar triturar las partículas duras, aunque los aglomerados blandos deberán desmenuzarse con una débil presión. Si el polvo es difícil de mojar, añadirle mojante en pequeñas cantidades hasta que se obtenga un mojado satisfactorio, pero emplear la mínima cantidad para evitar la formación de espuma durante el tamizado.
10.4.2 Extracción preliminar de las partículas finas.‒Utilizar tamices de unos 10 cm con los números de mallas requeridos por la especificación 10.6.1, introduciéndolos en agua y asegurarse de que la tela metálica esté completamente mojada; añadir mojante si fuera necesario. Sacar los tamices y emplearlos mientras estén mojados.
Diluir la pasta con agua hasta unos 150 ml y verterla sobre el tamiz más fino (10.6.2), enjuagando el vaso con agua. Lavar con un chorro suave de agua corriente, desechando cualquier producto que pase por el tamiz, hasta que el material retenido quede libre de partículas finas; cualquier agregado blando, que se pueda desmenuzar por una suave presión con la punta de los dedos o con la varilla de vidrio con el protector de goma, se dispersará de este modo.
10.4.3 Tamizado.‒Colocar el juego de tamices en orden, según el número de malla, siendo el superior el de paso más grueso. Transferir al tamiz superior el residuo del tamizado previo, lavar hasta que la cantidad de residuo del tamiz superior sea constante.
Quitar el tamiz superior, colocarlo sobre una cubeta y continuar llevando el producto dos minutos más. Transferir el producto que pasa por este tamiz al siguiente de la serie y repetir el lavado del tamiz más grueso durante dos minutos. Transferir de nuevo el producto que ha pasado al siguiente tamiz y repetir hasta que no pase nada en dos minutos (el punto final se estima más fácilmente en el tamizado húmedo que en el seco).
Repetir con cada tamiz sucesivo la técnica descrita para el tamiz más grueso, excepto para el producto que pase por el tamiz más fino que se desecha. Transferir cada residuo a un vidrio de reloj tarado, con la ayuda del chorro de agua destilada de un frasco lavador. Dejar que las partículas sedimenten y decantar la mayor parte del agua. Evaporar el resto a sequedad en baño de agua, completando el secado a 100 °C (o a otra temperatura según lo requieran sus propiedades físicas) y pesar el residuo.
10.5 Cálculo.
En el caso de ensayos con un solo tamiz, expresar el peso del residuo como porcentaje del peso de la muestra y registrar el resultado como porcentaje de retención para el tamiz de ensayo correspondiente.
Cuando haya que especificar en más de un tamiz, añadir al porcentaje del residuo de cada tamiz los porcentajes de residuos calculados de igual modo, de todos los tamices más gruesos de la serie. Anotar el total como porcentaje de retención al tamiz determinado.
10.6 Observaciones.
10.6.1 Como 9.5.1.
10.6.2 Como 9.5.2.
10.6.3 Como 9.5.3.
10.6.4 Como 9.5.4.
10.7 Referencias.
1. Collaborative International Pesticides Analytical Council Limited (CIPAC). Ed. 1970. MT/59.3.
11. DENSIDAD APARENTE DE PRODUCTOS EN POLVO
(Previa compactación)
11.1 Principio.
La densidad aparente del producto compactado es la que adquiere una determinada cantidad de polvo contenida en un recipiente normalizado, cuando éste se somete a vibración o compactación en condiciones específicas.
11.2 Material y aparatos.
11.2.1 Soporte de madera firmemente sujeto con tornillos a una superficie rígida (fig. 11.1).
11.2.2 Probeta de vidrio sin pico, provista de tapón de goma; la base de la probeta debe ser plana y el peso de la probeta con el tapón de goma debe ser de 250 ± 5 g. La probeta deberá estar graduada en divisiones de 2 ml, abarcando la escala de 25 a 250 ml. La distancia entre el 0 y el 250 de la probeta deberá ser de 22 a 24 cm. La elevación total de la probeta deberá ajustarse a los 25±2 mm mediante la aplicación a la parte inferior del tope de un suplemento adecuado.
11.2.3 Amohadilla para la base, de goma, de dureza Brithis Standard 35 a 50 o equivalente.
11.2.4 Cronómetro.
11.2.5 Balanza de platillos de fácil acceso que permita una precisión de 0,25 g.
11.2.6 Papel negro y satinado.
11.2.7 Dediles de goma suave.
11.2.8 Tamiz de 500 μ.
11.3 Procedimiento.
11.3.1 Preparación de la muestra (si se quiere).
Pesar aproximadamente 42 g de la muestra en un trozo de papel.
Tomar un papel de ensayo negro y satinado (de 25 x 25 centímetros), hacer dos pliegues paralelos formando un canal de 13 mm de ancho en su parte media y por su cara satinada. Poner el papel en la balanza y equilibrarla con otra hoja similar. Situar el papel anteriormente preparado sobre la mesa.
Colocar el tamiz sobre el papel, de manera que haya 5 cm de separación entre la tela metálica y el mismo.
Verter el polvo sobre el tamiz y frotar suavemente con los dedos cubiertos con dediles de goma. Si el tamiz tiende a obstruirse, levantarlo unos 25 mm y golpear suavemente los bordes con los dedos (pero no contra la mesa).
Poner el papel con la muestra tamizada en uno de los platillos, estando en el otro la hoja de contrapeso, junto con 40 g en pesas. Ajustar el peso de la muestra hasta equilibrarlo.
11.3.2 Determinación.‒Tomar 40 g de la muestra tamizada sobre el papel y colocarlo en posición inclinada. Con la palma de la mano coger el papel situándolo entre el pulgar y los otros dedos e introducirlo unos 13 mm en la probeta, que se ajustará con la otra mano formando un ángulo de 45 grados, aproximadamente, con la horizontal. Deslizar suavemente la muestra en la probeta. Si se introdujeran adherencias se evitarán dando suaves golpecitos con un dedo al final de la parte inferior del papel inclinado. La probeta no se golpeará ni sacudirá de ningún modo, y mientras se la está llenando no se hará ninguna presión sobre el polvo del papel.
Colocar el tapón de goma en la probeta sin dar sacudidas; introducir con cuidado ésta dentro del soporte de madera y empezar a cronometrar.
Con el pulgar y el índice de una mano sujetar con cuidado la parte superior de la probeta y, durante un segundo, levantarla en toda la extensión de su recorrido. Evitar que en el tope superior se produzca cualquier impacto indebido, para que al polvo no se le den sacudidas. Al principio del siguiente segundo, soltar la probeta quitando rápidamente y por completo el pulgar y el índice. Continuar este proceso de levantar y dejar caer la probeta con una frecuencia de dos segundos, hasta que se hayan contado 50 caídas. Cada vez que se levante la probeta se le debe girar unos 10 grados, con ello se conseguirá que se homogeneice la superficie superior, facilitando así la lectura del volumen final.
Al finalizar las 50 caídas, sacar inmediatamente la probeta del soporte de madera, ponerlo a la altura de la vista y acotar el volumen ajustándolo a los ml más próximos (V ml). No debe tomarse en consideración cualquier posterior descenso del nivel.
11.4 Cálculo.
Calcular la densidad aparente de la muestra (D g/ml), con dos cifras significativas, mediante la fórmula:

11.5 Referencias.
1. Collaborative International Pesticides Analytical Council Limited (CIPAC). Ed. 1970. MT/33.
12. DENSIDAD APARENTE DE PRODUCTOS EN GRÁNULOS
(Previa compactación sin presión)
12.1 Principio.
La densidad aparente del producto compactado es la que adquiere una determinada cantidad de gránulo contenido en un recipiente normalizado cuando éste se somete a vibración o compactación en condiciones específicas.
12.2 Material y aparatos.
12.2.1 Como en 11.2.1.
12.2.2 Como en 11 2.2.
12.2.3 Como en 11.2.3.
12.2.4 Como en 11,2.4.
12.2.5 Como en 11.2.5.
12.2.6 Como en 11.2.6.
12.2.7 Como en 11.2.7.
12.2.8 Como en 11.2.8.
12.3 Procedimiento.
Pesar en un vaso 40 g de la muestra e introducir ésta con cuidado en el interior de la probeta, acoplando el tapón de goma sin dar sacudidas. Colocarla con cuidado en el soporte de madera y empezar a cronometrar.
Con el pulgar y el índice de una mano, sujetar con cuidado la parte superior de la probeta y, durante un segundo, levantarla en toda la extensión de su recorrido. Evitar que en el tope superior se produzca cualquier impacto indebido para que a la muestra no se le den sacudidas.
Al principio del siguiente segundo, soltar la probeta quitando rápidamente y por completo el pulgar y el índice.
Continuar este proceso de levantar y dejar caer la probeta con una frecuencia de dos segundos hasta que se hayan contado 50 caídas. Cada vez que se levante la probeta se debe girar unos 10 grados: con ello se conseguirá que se homogeneice la superficie superior facilitando la lectura del volumen final. Al finalizar las 50 caídas, sacar inmediatamente la probeta del soporte de madera y ponerla a la altura de la vista y anotar el volumen, ajustándolo a los ml más próximos (V ml). No debe tomarse en consideración cualquier posterior descenso del nivel.
12.4 Cálculo.
Calcular la densidad aparente de la muestra (D g/ml), con dos cifras significativas, mediante la fórmula:
12.5 Referencias.
1. Collaborative International Pesticides Analytical Council Limited (CIPAC). Ed. 1970. MT/58.4.
13. ESTABILIDAD A BAJA TEMPERATURA
13.1 Principio.
La muestra se mantiene a 0 °C durante una hora y se anota el volumen de cualquier sustancia sólida o aceitosa que se haya separado. Continuar el almacenamiento a 0 °C durante siete días y por centrifugación hacer depositar las sustancias sólidas y aceitosas anotando su volumen.
13.2 Material y aparatos.
13.2.1 Refrigerador capaz de mantener la temperatura a 0 ± 1 ºC.
13.2.2 Tubos de centrífuga de fondo cónico de 100 ml de capacidad (fig. 13.I).
13.2.3 Centrífuga que proporcione una fuerza centrífuga relativa en el extremo de los tubos de 500 a 600 G.
13.2.4 Pipeta de 100 ml de capacidad.
13.3 Procedimiento.
13.3.1 Para concentrados emulsionables y disoluciones.‒Poner con exactitud de ± 1 ml 100 ml de una muestra del producto en un tubo de centrífuga. Enfriar el tubo y su contenido a 0 ± 1 °C en el refrigerador. Si el plaguicida de la muestra es un producto cristalino, añadir al tubo un pequeño cristal del pesticida de grado técnico o puro (ver 13.5.1).
Dejar que el tubo y su contenido estén a 0 ± 1 °C durante una hora. Durante este tiempo, agitar el contenido del tubo a intervalos de quince minutos, haciéndolo, cada vez, durante treinta segundos, aproximadamente. Después de este período, examinar el tubo y anotar el volumen de cualquier sustancia sólida o aceitosa presente. Volver a poner el tubo en el refrigerador y dejarle a 0 ± 1 °C durante un periodo de siete días.
Transcurridos los siete días, sacar el tubo del refrigerador y dejarlo reposar durante tres horas a temperatura ambiente, sin que llegue a sobrepasar 20 °C Tapar e invertir una vez el tubo, destapar y centrifugar durante quince minutos a una velocidad tal que la fuerza centrífuga relativa en el extremo de los tubos sea de 500 a 600 x G (aceleración debida a la gravedad: 981 cm/seg2).
13.3.2 Dilución acuosa.‒Poner 100 ml de la sustancia en el tubo de centrífuga e introducirlo en el refrigerador durante cuarenta y ocho horas a 0 ± 1 °C. Transcurrido este tiempo, anotar la cantidad de cualquier sustancia separada, si la hubiere. Dejar que el tubo de centrífuga adquiera la temperatura ambiente y anotar de nuevo la cantidad de sustancia separada.
13.4 Cálculo.
Anotar el volumen de cada una de las fases separadas, con una aproximación de 0,05 ml.
13.5 Observaciones.
13.5.1 El cristal de siembra debe ser obtenido de la muestra que se examina.
13.6 Referencias.
1. Collaborative International Pesticides Analytical Council Limited (CIPAC). Ed. 1970. MT/39.
14. FLUIDEZ DE LOS PRODUCTOS EN POLVO
14.1 Principio.
El método es adecuado para la determinación de la fluidez de los productos en polvo, como por ejemplo, cargas minerales, formulaciones en polvo (espolvoreables y mojables), etc. Los productos a ensayar deben pasar a través del tamiz de 250 μ.
En los productos cuyo tamaño de partícula sea mayor se apreciará la fluidez por inspección visual.
14.2 Material y aparatos.
14.2.1 Embudo de material adecuado, aluminio o similar de acuerdo con la descripción de la figura 14.I.
14.2.2 Frasco de vidrio con tapón a rosca de 100 ml de capacidad.
14.2.3 Tamices de 250 μ y 150 μ.
14.3 Reactivos.
14.3.1 Arena silícea limpia, seca y que pase por el tamiz de 250 μ, pero no por el de 150 μ.
14.4 Procedimiento.
Verter de 10 a 15 g de la muestra en el embudo estándar, golpear suavemente éste una o dos veces si hiciera falta y observar si fluye. Si esto ocurre la fluidez es «0».
Si no fluye, pesar 5 g con aproximación de 0,1 g de la muestra en el frasco (14.2.3), añadir 5 ± 0,1 g de arena y mezclar a mano durante cinco minutos por lo menos. Pasar la mezcla cuidadosamente al embudo, golpear suavemente éste una o dos veces si hiciera falta y observar si la mezcla fluye, si la mezcla no fluye a través del orificio, volverla a pasar al frasco y mezclarla con otros 5 ± 0,1 g de arena. Repetir el procedimiento añadiendo la arena en porciones de 5 ± 0,1 g hasta que fluya.
14.5 Cálculo.
Si la muestra fluye libremente, considerar como índice de fluidez «0». Si no, considerar como índice de fluidez el número mínimo de veces que ha sido preciso añadir porciones de 5 ± 0,1 gramos de arena a los 5 g de muestra para conseguir que fluya por lo menos durante quince segundos.
14.6 Observaciones.
14.6.1 Índice de fluidez es el número mínimo de partes en peso de arena silícea que tiene que añadirse a una parte en peso de la muestra para que fluya.
14.6.2 Fluidez.‒Se dice que una sustancia «fluye» cuando sale libremente en chorro continuo, por un embudo estándar durante quince segundos como mínimo.
14.6.3 Cuando exista posibilidad de cargas electrostáticas el frasco de vidrio debe ser sustituido por un frasco metálico con tapón de rosca.
Fig. 14.1
14.7 Referencias.
1. Collaborative International. Pesticides Analytical Council Limited (CIPAC). Ed. 1970. MT/44.
15(a). PUNTO DE INFLAMACIÓN EN VASO CERRADO
(Método Pensky-Martens)
15(a).1 Principio.
Determinar la temperatura más baja a la cual, al aplicar una llama, se inflama el vapor formado sobre la muestra, previamente calentada uniformemente.
Aplicable a productos cuyo punto de inflamación es superior a 48 ºC.
15(a).2 Material y aparatos.
15(a).2.1 Aparato Pensky-Martens.
15(a).2.2 Termómetros adecuados para lecturas entre ‒7 y 93 ºC; 93 y 110 ºC; 110 y 370 ºC.
15(a).3 Procedimiento.
15(a).3.1 Sustancias que no sean suspensiones de sólidos.
Limpiar y secar perfectamente todas las partes del vaso y sus accesorios antes de empezar el ensayo. Poner especial cuidado en evitar el dejar en el aparato restos de disolvente empleado para limpiarlo tras el ensayo anterior. Llenar el vaso con la muestra a ensayar hasta el enrase. Colocar la tapa sobre el vaso y ponerlo en el baño de aire cuidando de que estén perfectamente encajadas las muescas semicirculares del aparato. Insertar el termómetro adecuado. Si se conoce la temperatura aproximada a que se inflamará la muestra, utilizar directamente el termómetro apropiado. En caso contrario utilizar la serie de termómetros. Encender la llama y graduarla al tamaño de una perla de 4 mm de diámetro. Calentar a una velocidad tal que la temperatura leída en el termómetro no aumente menos de 5 °C, ni más de 6 °C por minuto. Agitar a razón de 90 a 120 r.p.m.
Aplicar por primera vez la llama cuando la temperatura sea de unos 16 °C por debajo del punto de inflamación esperado. Aplicar la llama a cada aumento de 1 °C, hasta alcanzar los 100 °C, y después hacerlo a cada aumento de 3 °C. Aplicar la llama operando con el aparato que controla el obturador y el mechero con la llama del ensayo, de forma que la llama baje en 0,5 segundos, manteniéndola durante un segundo en su posición más baja y elevarla rápidamente a su posición más alta. No agitar mientras se aplica la llama.
Tomar como punto de inflamación la temperatura que marque el termómetro en el momento en que la aplicación de la llama origine una clara inflamación en el interior del vaso. No confundir el verdadero punto de inflamación con el halo azulado que a veces rodea a la llama de ensayo, en las aplicaciones que preceden a la que origina la verdadera inflamación.
Observar y anotar la presión barométrica. La cifra del punto de inflamación se corregirá sumando o restando 0,9 °C por cada 25 mm de Hg en que la presión barométrica esté por debajo o por encima de los 760 mm de Hg.
15(a).3.2 Suspensiones de sólidos.
Llevar la sustancia a ensayar y el aparato a una temperatura de 10 a 20 °C. Llenar completamente el espacio que hay entre el vaso y el baño de aire, con agua a la misma temperatura del aparato y de la muestra. Agitar a razón de 250 ± 10 r.p.m. Durante el ensayo la temperatura debe ir aumentando a una velocidad comprendida entre 1 y 2 °C por minuto. Continuar como en 15(a).3.1.
15(a).4 Cálculo.
Expresar los resultados como punto de inflamación con aparato Pensky-Martens.
Los resultados de los ensayos duplicados no deben diferir en más de las siguientes cantidades:
| Punto de inflamación | Repetición | Reproducibilidad |
|---|---|---|
| Suspensiones de sólidos. | 2,2 °C | 3,3 °C |
| 104 °C e inferiores. | 2,2 °C | 3,3 °C |
| Superiores a 104 °C. | 5,5 °C | 8,3 °C |
15(a).5 Referencias.
1. Collaborative International Pesticides Analytical Council Limited (CIPAC). Ed. 1970. MT/12.3.
15(b). PUNTO DE INFLAMACIÓN EN VASO CERRADO
(Método Tag)
15(b).1 Principio.
Determinar la temperatura más baja a la cual al aplicar una llama se inflama el vapor formado sobre la muestra, previamente calentada uniformemente. Aplicable a productos cuyo punto de inflamación es inferior a 79 °C.
15(b).2 Material y aparatos.
15(b).2.1 Aparato de ensayo Tag.
15(b).2.2 Una pantalla protectora de base cuadrada de 46 cm de lado por 51 cm de altura, abierta por delante.
15(b).2.3 Termómetros adecuados para el vaso de ensayo y el baño.
15(b).3 Procedimiento.
Realizar el ensayo con luz tenue para observar con claridad la inflamación. Dicha determinación no debe hacerse en vitrina o cerca de ventiladores, ya que darían resultados dudosos.
Colocar el aparato, fijo y nivelado; poner el termómetro del baño en su sitio; poner un recipiente debajo del tubo aliviadero para recoger el excedente, y llenar el baño con agua a una temperatura tal que, cuando empiece el ensayo, la temperatura del baño de agua esté al menos 11 °C por debajo del probable punto de inflamación de la muestra a ensayar.
Poner el vaso vacío en el baño de agua; medir 50 ml del producto a ensayar en una probeta y verterlo en el vaso.
Eliminar en la superficie de la muestra cualquier burbuja de aire que pueda existir empleando para ello un trozo de papel limpio y seco.
15(b).3.1 Si se dispone de gas.‒Tapar el vaso colocando en su sitio el termómetro que medirá el punto de inflamación; encender el mechero de gas de la tapa, regulando su llama con la válvula de gas hasta hacerla del tamaño de la pequeña perla blanca de la tapa.
15(b).3.2 Si no se dispone de gas.‒Después de cerrar la válvula, insertar una mecha de algodón en la boquilla del mechero, poner una pequeña cantidad de hilacha de algodón en el depósito de aceite y llenarlo con aceite de ballena, grasa de cerdo (o petróleo). Colocar el tapón del depósito de combustible, pero no enroscarlo muy fuerte, ya que conviene dejar una pequeña abertura para la entrada de aire.
Encender el mechero lleno de alcohol, colocarlo en la base del aparato y ver que esté situado en el centro. Graduar la llama de la lámpara de alcohol para que la temperatura de la muestra que está en el vaso suba a razón de 1 ºC por minuto ‒no más rápidamente de 1,1 °C ni menos de 0,9 °C por minuto. El mechero de gas puede sustituir al de alcohol sin que afecte apreciablemente al resultado del ensayo.
Al comenzar la prueba, anotar la presión barométrica así como la temperatura de la muestra.
Cuando la temperatura de la muestra llega a unos 5 ºC por debajo del punto de inflamación probable, se hace girar el botón de la tapa, para introducir la llama dentro del vaso y volverla a poner en su posición primitiva, rápida pero no bruscamente. El tiempo de esta operación debe ser aproximadamente de un segundo.
Anotar la temperatura de la muestra y la hora en que se hizo la primera introducción de la llama.
Repetir la aplicación de la llama a cada aumento de 9,6 °C de temperatura de la muestra, hasta que se produzca la inflamación de ésta dentro del vaso. No hay que confundir la inflamación con el ensanchamiento de la llama de ensayo o con el halo que forma ésta cuando se introduce dentro del vaso; la verdadera inflamación consume el gas de la parte superior del vaso y causa una ligera explosión. (Si el aumento de la temperatura de la muestra, desde el momento que se introduce por primera vez la llama, hasta que se alcanza el punto de inflamación, es mayor que 1,1 °C o menor que 0,9 °C por minuto, se deberá considerar dudoso el ensayo y se ajustará la lámpara de alcohol o el mechero de gas para corregir la velocidad de calentamiento.)
Anotar el punto de inflamación y la hora en que se alcanza.
No apagar la llama de ensayo con la válvula reguladora. Dejar ésta ajustada para obtener el tamaño adecuado de llama.
Una vez realizado por completo este ensayo preliminar, retirar la lámpara de alcohol o mechero de gas. Levantar la tapa del vaso y limpiar el termómetro. Se saca el vaso, se vacía y se seca cuidadosamente. Tirar la muestra una vez se haya analizado.
Verter agua fría dentro del baño de agua, dejando que rebose hacia el receptáculo hasta que la temperatura del baño de agua descienda a 8 ºC por debajo del punto de inflamación de la muestra, obtenido en el ensayo anterior.
Colocar en el baño de agua el vaso con 50 ml de muestra nueva. Eliminar de la superficie de la muestra cualquier burbuja, poner la tapa con su termómetro, aplicar la lámpara de alcohol o el mechero de gas; anotar la temperatura de la muestra y la del agua, y repetir el ensayo antes descrito. Introducir por primera vez la llama cuando la temperatura sea 5,5 ºC inferior al punto de inflamación obtenido en el ensayo anterior.
Si los resultados de dos determinaciones difieren en más de 0,6 °C, realizar una tercera determinación y si la máxima variación de los tres ensayos no es superior a 1,1 °C, se tomará como punto de inflamación el promedio de las tres, corregido en función de la presión barométrica.
15(b).4 Cálculo.
Leer y anotar la presión barométrica.‒Cuando las lecturas del barómetro varíen en más de 13 mm de Hg de la presión normal de 760 mm de Hg, el valor del punto de inflamación se corregirá de acuerdo con la tabla 15(b).1, que ha sido calculada considerando una diferencia de 0,9 °C para cada variación de 25 mm en la presión barométrica.
TABLA 15(b).I
Corrección del punto de inflamación con la presión barométrica
|
P. barométrica (mm Hg) |
Corrección (°C) |
|---|---|
| 700 | + 2,2 |
| 705 | + 2,0 |
| 710 | + 1,8 |
| 715 | + 1,6 |
| 720 | + 1,4 |
| 725 | + 1,3 |
| 730 | + 1,1 |
| 735 | + 0,9 |
| 740 | + 0,7 |
| 745 | + 0,5 |
| 750 | + 0,4 |
| 755 | + 0,2 |
| 760 | + 0 |
| 765 | ‒ 0,2 |
| 770 | ‒ 0,4 |
| 775 | ‒ 0,5 |
| 780 | ‒ 0,7 |
| 785 | ‒ 0,9 |
15(b).5 Referencias.
1. Collaborative International Pesticides Analytical Council Limited (CIPAC). Ed. 1970. MT/12.2.
16. MOJABILIDAD DE POLVOS DISPERSABLES
(Mojables)
16.1 Principio.
Determinación del tiempo empleado para lograr el completo mojado de los polvos en agua.
16.2 Material y aparatos.
16.2.1 Vaso de 250 ml de 6,5 ± 0,5 cm de diámetro interno y de 0,9 ± 0,5 cm de altura.
16.2.2 Pesasustancias.
16.2.3 Cronómetro.
16.2.4 Probeta de 100 ml.
16.3 Reactivos.
16.3.1 Agua patrón como en 1.3.1.
16.4 Procedimiento.
16.4.1 Sin agitación.‒Poner 100 ± 1 ml de agua patrón en el vaso. Pesar 5 ± 0,1 g de una muestra representativa del polvo, teniendo cuidado de que no se compacte. Añadir de una vez todo el polvo, dejándolo caer en el agua desde el borde del vaso, y sin mover para nada el vaso, con objeto de que no se produzcan agitaciones en la superficie del líquido.
Una vez añadido el polvo empezar a cronometrar y anotar el tiempo transcurrido (con aproximación de segundo) hasta que esté completamente mojado (debe despreciarse la película de partículas que permanecen en la superficie).
16.4.2 Con agitación.‒Realizar la técnica descrita en 16.4.1, pero el contenido del vaso debe agitarse a mano de forma rotacional con una frecuencia de 120 rotaciones por minuto tras la adición del polvo. Referir el resultado como «tiempo de mojado con agitación».
16.5 Referencias.
1. Collaborative International Pesticides Analytical Council Limited (CIPAC). Ed. 1970. MT/53.3.
MANTEQUILLA
9. ÁCIDOS GRASOS DE CADENA CORTA
9.1 Principio.
Obtención de los ésteres metílicos de los ácidos grasos mediante reacción con una solución de hidróxido potásico en metanol y subsiguiente inyección directamente de la disolución de ésteres metílicos en el cromatógrafo.
El método es aplicable a las grasas de mantequillas u otras que contengan ácidos grasos de longitud de cadena inferior al C14 y siempre que el contenido de ácidos libres no exceda del 1 por 100 expresados en ácido oleico.
9.2 Material y aparatos.
9.2.1 Matraces con boca esmerilada y fondo redondo de 50 y 100 ml de capacidad.
9.2.2 Pipetas aforadas de 1, 2 y 10 ml.
9.2.3 Matraces aforados de 50 y 100 ml de capacidad.
9.2.4 Probeta graduada de 10 ml.
9.2.5 Jeringa de características adecuadas para la inyección de la muestra, graduada en décimas de ml, con una capacidad total de 1 a 10 ml.
9.2.6 Cromatógrafo apto para trabajar en fase gaseosa, provisto de horno capaz de ser calentado hasta 250-300 °C y sistema de regulación que permita controlar la temperatura con un error de ± 1,0 °C. Equipado con programador de temperatura capaz de llevar la temperatura del horno de 60 °C a 180 °C a una velocidad de 4 °C/min. Provisto de regulación independiente de la temperatura del inyector, que podrá ser calentado a una temperatura superior por lo menos en 50° a la máxima alcanzable por el horno provisto de un sistema de detección sensible, de ionización de llama de hidrógeno, que pueda ser mantenido a la temperatura de la columna, a unos 50 °C por encima de la del horno.
9.2.7 Registrador con una tensión de entrada adecuada a la salida del amplificador del cromatógrafo, con una velocidad de respuesta mínima capaz de producir la deflexión completa de la escala en un segundo; y una velocidad de desplazamiento del papel de 5 mm/min, que permita la posibilidad de variar esta velocidad, acelerando o retardando el desplazamiento.
9.2.8 Tubo de nitrógeno a presión, utilizable como gas portador, debiendo tener una riqueza mínima del 99,8 por 100.
9.2.9 Tubos de hidrógeno y aire a presión necesarios para el caso en que se utilice detector de llama de hidrógeno. El hidrógeno deberá tener una riqueza mínima del 99,8 por 100, debiendo estar seco. Como medida de seguridad, es muy conveniente colocar a la entrada de los gases en el cromatógrafo, sendos tubos de desecación, provistos de tamiz molecular 13X.
9.2.10 Columna cromatográfica.
9.2.10.1 Columna que satisfaga las condiciones que se indican en 9.6.1.
9.2.10.2 Columna de vidrio con diámetro interior de 4 mm y una longitud aproximada de 2 mm. Rellenada con Chromosorb G, W o Q (80-100 mallas), conteniendo de 2,5 a 5 por 100 de un poliéster, recomendándose cualquiera de los tres siguientes: dietilenglicolsuccinato (DEGS); etilenglicolsuccinato o adipato (EGS o EGA): polietilenglicoladipato (PEGA).
Antes de emplear una columna nueva en la resolución de problemas analíticos, debe ser acondicionada, eliminando todos aquellos productos volátiles que perturbarían la marcha de la cromatografía. Para ello, se monta en el cromatógrafo, sin conectarla al detector, y se calienta el horno a unos 10 °C por encima de la temperatura máxima a que vaya a ser utilizada la columna en trabajos posteriores; haciendo pasar, al mismo tiempo, una corriente de nitrógeno de 30 a 40 ml/minuto, que se mantiene durante veinticuatro horas como mínimo. La columna será apta para su utilización si, una vez conectada al detector y en funcionamiento normal, la línea base dibujada por el registrador acusa la estabilidad del sistema.
9.3 Reactivos.
9.3.1 Metanol absoluto (99,8 por 100).
9.3.2 Hidróxido potásico, en lentejas.
9.3.3 Éter de petróleo o hexano. Éter de petróleo (p. e.: 40-60 °C), cuyo contenido en benceno no sea superior a 0,1 por 100. Hexano normal, que cumpla las mismas especificaciones del éter de petróleo.
9.3.4 Heptano normal, con una riqueza mínima del 99 por 100.
9.3.5 Disolución 2 N de hidróxido potásico en metanol. Disolver 11,2 g de hidróxido potásico en 100 ml de metanol.
9.3.6 Ésteres metílicos de pureza adecuada para su utilización como patrones de cromatografía gaseosa.‒Se dispondrá de los ésteres metílicos de los ácidos mencionados a continuación, debiendo tener una pureza mínima de 99 por 100, determinada por cromatografía gaseosa:
Ácido butanoico (butírico).
Ácido pentanoico (valeriánico).
Ácido hexanoico (caproico).
Ácido octanoico (caprílico).
Ácido decanoico (cáprico).
Ácido dodecanoico (láurico).
Ácido tetradecanoico (mirístico).
Ácido hexadecanoico (palmítico).
Ácido octadecanoico (esteárico).
Ácido 9-octadecanoico (oleico).
Ácido 9,12 octadecadienoico (linoléico).
Ácido sicosanoico (aráquico).
9.3.7 Solución de referencia I.‒En un matraz aforado de 50 ml se pesa, con exactitud de ± 0,1 mg, 1 g de pentanoato de metilo, disolviéndolo en heptano normal y completando hasta el enrase.
9.3.8 Solución de referencia II.‒En un matraz aforado de 100 ml se pesa, con exactitud de ± 0,1 mg, 200 g de pentanoato de metilo, disolviéndolo en heptano normal y completando hasta el enrase.
9.4 Procedimiento.
9.4.1 Preparación de los ésteres metílicos.
En un matraz de fondo redondo de 50 ml, pesar con exactitud de ± 0,1 mg, 1 g de grasa. Añadir 10 ml de éter de petróleo o hexano y agitar suavemente hasta disolución de la grasa.
En el caso de que se quiera efectuar una determinación cuantitativa de los ácidos butírico y caproico en la muestra, agregar a la disolución en éter de petróleo de la grasa 1 ml, exactamente medido, de la solución de referencia más adecuada; para muestras conteniendo de 1-4 por 100 de ácido butírico se utilizará la solución de referencia I; para muestras conteniendo menos de 1 por 100 de ácido butírico se utilizará la solución de referencia II.
Si se desea efectuar solamente un análisis completo de la fracción de ácidos grasos, para lo que se aplica el método de normalización interna, no será necesario el empleo de solución de referencia.
A la solución en éter de petróleo de la muestra, adicionada o no de solución de referencia, agregar 0,5 ml de disolución 2 N de hidróxido potásico. Agitar suavemente la mezcla hasta que se ponga transparente, para lo cual son suficientes unos veinte a treinta segundos. Casi inmediatamente después de observar la clarificación de la solución suele apreciarse un enturbiamiento debido a la separación de glicerol, que se sedimenta rápidamente.
Inmediatamente después de terminada la reacción y observada la sedimentación, tomar la cantidad necesaria con la jeringa e inyectar en el cromatógrafo; una demora en la inyección de los ésteres metílicos daría lugar a la formación de jabones, con error en la determinación.
9.4.2 Determinación cromatográfica.
9.4.2.1 Condiciones de trabajo.
Temperatura de la columna: Temperatura programada de 60 °C a 160 °C, con una velocidad de 4 ºC/minuto.
Temperatura del inyector: 200 °C.
Temperatura del detector: 200 °C.
Gas portador: Nitrógeno (o helio) con un flujo de 60 ml/min.
Flujo de hidrógeno y aire para la alimentación del detector: Los flujos dependerán del tipo de detector utilizado, debiendo determinarse previamente para optimizar la respuesta.
El registro obtenido del cromatograma debe satisfacer las condiciones que se indican a continuación; caso contrario, se repite la inyección modificando la cantidad inyectada o la sensibilidad de trabajo hasta obtener un cromatograma satisfactorio.
Los requisitos exigidos son los siguientes:
a) El área total descrita en el registro, referida a la sensibilidad máxima utilizada en el curso de la operación, debe ser de un orden aproximado de 2.000 mm2, con una velocidad del papel en el registrador de 5 mm/min. De esta forma, los componentes presentes en una cuantía del 0,1 por 100 deben dar un pico, como mínimo, de 2 mm2, siendo, por tanto, perfectamente reconocibles.
b) Con el fin de conseguir que todos los picos caigan dentro del papel registrador, se utilizará, en cada caso, la atenuación de sensibilidad que sea necesaria, cuidando que el pico de mayor intensidad no sea atenuado más de ocho veces.
Una vez conseguido un registro satisfactorio, y habiendo alcanzado nuevamente la pluma la línea base, se interrumpe el funcionamiento del registrador y se retira el papel con el registro para la identificación de los picos y/o cálculos cuantitativos.
9.4.2.2 Identificación de los picos.‒Se seguirán los criterios establecidos en el apartado 9.6.2.
9.4.2.3 Determinaciones cuantitativas.‒La determinación cuantitativa se basa en el principio de que los pesos de cada uno de los componentes separados en la mezcla son proporcionales a las áreas comprendidas dentro de los triángulos dibujados debajo de cada pico. El área de cada triángulo se obtiene trazando rectas tangentes a las líneas dibujadas en el registro, prolongándolas hasta su intersección con la línea base y multiplicando la altura del triángulo por la mitad de la base. En el caso de haber trabajado con atenuaciones diferentes para cada pico, se referirán todas las medidas a una misma sensibilidad del registrador, multiplicando la altura por el factor de atenuación correspondiente en cada caso, y el valor de la altura así corregida por la mitad de la base.
9.4.3 Determinación del contenido de los ácidos butírico y caproico en la materia grasa.‒Esta determinación se realiza por el método del patrón interno, siendo el patrón elegido el pentanoato de metilo.
9.4.3.1 Preparación de la mezcla de calibración.‒Con una exactitud de ± 0,1 mg y en un matraz aforado de 50 ml, pesar unos 100 mg de cada uno de los siguientes patrones: butanoato de metilo, pentanoato de metilo y caproato de metilo. Se disuelve la mezcla de heptano normal y se diluye completando hasta el enrase.
Inyectar la cantidad necesaria de la solución anterior, normalmente 0,2-0,4 μl, para que, trabajando a la sensibilidad media del aparato, se consiga situar los máximos de los picos en una posición del 70-80 por 100 del recorrido total de la pluma del registrador. Los tres picos deberán registrarse a la misma sensibilidad. Si fuese necesario, se diluirá la solución anterior con heptano normal en la relación necesaria para poder ajustarse a las prescripciones fijadas. Efectuar, cuando menos, tres determinaciones consecutivas, que no deben discrepar entre sí más del 1 por 100.
9.4.4 Análisis cuantitativo de la totalidad de los componentes de la fracción de ácidos grasos, comprendiendo del C4 al C20 y C18:3.
9.4.4.1 Preparación de la mezcla de calibración.‒Determinar previamente el factor de corrección para cada ácido componente de la mezcla, referido a uno cualquiera de ellos que se toma como patrón, eligiéndose normalmente para este fin el ácido palmítico, y debiendo tener la mezcla de calibración una composición análoga a la de la mezcla problema.
Para ello, si no se conoce previamente el orden de composición del problema, se realizará una determinación cromatográfica de orientación, realizándose en el registro la cuantificación de los componentes suponiendo el mismo factor de respuesta para todos ellos, efectuando un reparto proporcional entre las áreas medidas.
En un matraz aforado de 50 ml, pesar, con una exactitud de ± 0,1 mg, cantidades de los ésteres metílicos patrones que se indican a continuación proporcionales a las cifras de composición encontradas en el análisis de orientación anteriormente aludido, o previstas con anterioridad para la muestra. Los patrones que deben pesarse son los siguientes: butanoato de metilo; hexanoato de metilo, octanoato de metilo, decanoato de metilo, dodecanoato de metilo, tetradecanoato de metilo; hexadecanoato de metilo, octadecanoato de metilo, oleato de metilo, linoleato de metilo y eicosanoato de metilo. Se disuelve la mezcla de heptano normal agregando la cantidad adecuada de disolvente en relación al peso total de ésteres metílicos que se hayan pesado; para unos 500 mg en total, se deben emplear, como orientación, unos 50 ml de heptano. A continuación, inyectar 0,2-0,4 μl para que trabajando a la sensibilidad media del aparato, se consiga situar el máximo del pico correspondiente al componente mayoritario, en una posición del 70-80 por 100 del recorrido total de la pluma del registrador. Todos los picos deben registrarse a la misma sensibilidad, lo cual suele ser perfectamente factible en la grasa de leche; en aquellos casos en que la relación entre el pico mayoritario y el pico minoritario no permita registrar este último con las dimensiones adecuadas para efectuar una cuantificación correcta de su área, se podrá efectuar el cambio necesario en la atenuación del registro, procurando que ésta no sobrepase la relación de 4:1. Si fuese necesario, se diluirá la solución con heptano normal en la relación necesaria para poder ajustarse a las prescripciones fijadas. Efectuar, cuando menos, tres determinaciones consecutivas, que no deben discrepar entre sí más del 1 por 100.
9.5 Cálculos.
9.5.1 Cálculo de los factores de corrección para los ácidos butírico y caproico.‒Se determinan las áreas de los tres picos, siguiendo las normas que se contienen en el apartado 5.4, y se calculan los dos factores correspondientes al C4 y C6, con la fórmula siguiente:

Siendo:
fx = factor de corrección del ácido x.
x = cantidad pesada del ácido x.
Ap = área medida en el registro para el patrón de pentanoato.
Ax = área medida en el registro para el ácido x.
P = peso del patrón pentanoato.
9.5.2 Cálculo del contenido de ácidos.‒Los contenidos de ácido butírico y ácido caproico en la muestra de grasa se calculan por la fórmula siguiente:

fx = factor de corrección determinado para cada ácido, según se indica en el párrafo anterior.
Ax = área medida en el registro para el ácido.
P = peso del patrón interno (pentanoato).
Ap = Área medida en registro para el patrón interno.
M = peso de la muestra de grasa.
9.5.3 Cálculo de los factores de corrección.‒Una vez determinadas las áreas de todos los picos, siguiendo las normas que se contienen en el apartado 9.4.2.2, se calcula el factor de cada Ácido, referido al ácido palmítico tomado como unidad, utilizando la fórmula que se incluye en el apartado 9.5.1, sustituyendo el área Ap y el peso P del compuesto patrón por los valores correspondientes al palmitato de metilo.
9.5.4 Cálculo de composición de la fracción de ácidos grasos.‒Se calcularán las áreas corregidas de cada uno de los componentes de la fracción multiplicando el área medida en el registro por el factor de corrección determinado según se indica en el apartado anterior. El contenido de cada componente vendrá dado por la expresión:

Siendo:
fx = factor de corrección del componente x.
Ax = área medida en el registro para el componente x.
Σ (fx • Ax) = suma de todas las áreas corregidas correspondientes a los componentes de la fracción.
9.6 Observaciones.
9.6.1 La puesta a punto de la columna se determina obteniendo la resolución de dos productos críticos como son el oleato y el estearato de metilo. La resolución viene determinada por la expresión:

Siendo:
D = distancia entre los dos máximos de los picos del oleato y el estearato.
O = ancho de la base del pico correspondiente al oleato.
E = ancho de la base del pico correspondiente al estearato.
Estos valores se determinan sobre el cromatograma obtenido con una muestra conteniendo cantidades aproximadamente iguales de estearato y oleato de metilo, inyectando una cantidad tal que la altura de estos picos alcance al 25-50 por 100 del ancho del papel de registro. Si la resolución calculada es igual o mayor que 1,0, la columna y el instrumento se encuentran en condiciones satisfactorias. Todas las columnas en el transcurso de su utilización sufren una pérdida gradual en la resolución de los picos; cuando el valor llegue a ser inferior a 1,0, deberá instalarse una nueva columna.
9.6.2 Para la identificación de los picos se pueden seguir dos criterios:
9.6.2.1 Criterio basado en los tiempos de retención.‒Refiriéndonos exclusivamente a los ácidos que entran normalmente en la composición de las grasas naturales, sus ésteres aparecen en el cromatograma en orden creciente de sus átomos de carbono y a su insaturación. Esto es, el palmítico (C16) aparece delante del esteárico (C18), y los ésteres en C18 aparecen en el orden estearato, oleato, linoleato y linolenato. El éster del ácido aráquico (C20:0), usualmente, aparece antes del linolénico (C18:3), pero puede ocurrir lo contrario en algunos casos, dependiendo del tipo de columna y de las condiciones de su utilización, o incluso superponerse el uno al otro.
Operando en condiciones constantes, los tiempos de retención son reproductibles en cada especie química, siendo el criterio más frecuente empleado para su identificación.
El tiempo de retención viene dado por la distancia, medida en el cromatograma, entre el máximo del pico del aire y la posición del máximo de la banda. Trabajando con detector de llama de hidrógeno, la salida del aire no se detecta, pudiéndose tomar, en este caso, el momento en que se inicia la salida del disolvente, acusada por una fuerte deflexión de la pluma del registrador.
9.6.2.2 Criterio basado en los tiempos de retención relativos.‒Los tiempos de retención relativos son más reproductibles. Las retenciones relativas vienen determinadas por el cociente de dividir el tiempo de retención de cada pico por el tiempo registrado para el pico del palmitato de metilo, o bien por otro éster que se tome como comparación, determinados todos ellos según el criterio expuesto en el párrafo anterior.
9.7 Referencias.
1. Instituto de Racionalización y Normalización del Trabajo. Una Norma Española 55.118.
QUESO
1. EXTRACCIÓN DE LA GRASA DEL QUESO
1.1 Principio.
Extracción de la grasa del queso mediante pentano o éter de petróleo.
1.2 Material y aparatos.
1.2.1 Mortero.
1.2.2 Aparato de extracción continuo.
1.2.3 Baño de agua.
1.3 Reactivos.
1.3.1 Sulfato sódico anhidro.
1.3.2 Pentano o éter de petróleo (p. e. 40-60 °C).
1.4 Procedimiento.
Moler la muestra en un mortero con sulfato sódico anhidro hasta obtener una masa granulosa. Extraer la masa con pentano o éter de petróleo (se puede usar un aparato de extracción continuo) y evaporar el disolvente al baño de agua o a presión reducida.
1.5 Referencia.
1. Norma internacional FIL-IDF 32: 1965.
2. DETERMINACIÓN DEL CONTENIDO EN MATERIA GRASA
2.1 Principio.
El contenido de grasa se determina gravimétricamente por digestión del queso con ácido clorhídrico y subsiguientemente extracción de la grasa de una solución ácido-alcohólica con la ayuda de éter dietílico y éter de petróleo, evaporación de los disolventes y posterior pesada de los residuos.
La precisión del método es de 0,2 g de grasa por 100 g del producto.
2.2 Material y aparatos.
2.2.1 Balanza analítica.
2.2.2 Probetas o matraces de extracción adecuados provistos de tapones de vidrio esmerilado o corcho; dispositivos del cierre que no puedan ser atacados por los disolventes utilizados. Si se usan tapones de corcho deberán ser de buena calidad, sometiéndolos a extracción sucesivamente con éter dietílico y éter de petróleo. Después se introducirán, al menos durante veinte minutos, en agua a una temperatura de 60 °C o superior, dejándose enfriar en agua de forma que estén saturados cuando se utilicen.
2.2.3 Matraces de paredes delgadas y bases planas de 150 a 250 ml de capacidad.
2.2.4 Estufa de desecación regulable que permita trabajar a 102° ± 2 °C, o una estufa de desecación por vacío (temperatura de 70° a 75 °C, presión menor de 50 mm de Hg).
2.2.5 Perlas de vidrio o trozos de carburo de silicio, exento de grasa.
2.2.6 Baño de agua.
2.2.7 Hojas de película de celulosa, sin barnizar, solubles en ácido clorhídrico, de 0,03-0,05 mm de espesor y de 50 x 75 mm de superficie, aproximadamente. Las películas de celulosa no deben afectar al resultado del análisis.
2.2.8 Aparato adecuado para la trituración de la muestra.
2.3 Reactivos.
2.3.1 Ácido clorhídrico del 25 por 100 (p/p) (d20 = 1,125).
2.3.2 Etanol del 96 por 100 (v/v) (2.6.1).
2.3.3 Éter dietílico, exento de peróxidos (2.6.2).
2.3.4 Éter de petróleo que destile a una temperatura que oscile entre 30° y 60 °C.
2.3.5 La mezcla de disolventes se prepara poco antes de utilizarla, mezclando volúmenes iguales de 2.3.3 y 2.3.4 (2.6.3).
2.4 Procedimiento.
2.4.1 Preparación de la muestra.‒Antes de efectuar el análisis, eliminar la corteza, capa o superficie mohosa que recubre el queso, con objeto de obtener una muestra representativa del queso tal como se consume normalmente. Triturar la muestra con 2.2.8, mezclar la masa triturada rápidamente, y si es posible triturarla por segunda vez y mezclarla de nuevo concienzudamente (2.6.4). Pasar la muestra preparada a un recipiente cerrado herméticamente hasta el momento del análisis, que se efectuará en el mismo día (2.6.5).
2.4.2 Determinación.‒Secar el matraz 2.2.3 con 2.2.5 en la estufa durante un intervalo de media hora. Dejar que se enfríe el matraz a la temperatura ambiente de la balanza y pesar el matraz enfriado con aproximación de 0,1 mg.
Pesar, con aproximación de 1 mg en el aparato de extracción 2.2.2 o en un vaso o matraz de 100 ml, de 1 a 3 g de la muestra de queso preparada. La muestra del ensayo podrá también pesarse utilizando una lámina de celulosa 2.2.7, que posteriormente se plegará e introducirá en el tipo de vasija seleccionada.
Añadir de 8 a 10 ml de ácido clorhídrico (según la forma del aparato de extracción) y agitar la vasija ligeramente en un baño de agua hirviendo o sobre una llama hasta que el queso esté completamente disuelto. Dejar la vasija en reposo durante veinte minutos en el baño de agua hirviendo y después enfriar, por ejemplo, en agua corriente.
Si la digestión del queso se ha hecho en el aparato de extracción, añadir 10 ml de etanol y mezclar el contenido, removiéndolo ligeramente, pero de un modo homogéneo en el aparato sin cerrar.
Si la digestión del queso se ha hecho en una vasija distinta del matraz de extracción, verter el contenido de la vasija en este matraz. Enjuagarlo sucesivamente con 10 ml de etanol, 25 ml de éter dietílico y 25 ml de éter de petróleo, vertiendo cada vez el disolvente en el matraz de extracción. Después de cada adición mezclar y agitar el matraz de extracción, según se indica a continuación.
Añadir 25 ml de éter dietílico, cerrar el aparato y agitar vigorosamente, invirtiéndolo repetidamente durante un minuto. Enfriarlo, si es necesario, en agua corriente. Quitar el tapón cuidadosamente y añadir 25 ml de éter de petróleo, empleando los primeros ml para enjuagar el tapón y la superficie interna del cuello del aparato, dejando que el líquido de los enjuagues penetre en el mismo. Cerrarlo, volviendo a colocar el tapón, agitar e invertirlo repetidamente durante treinta segundos; no debe agitarse demasiado enérgicamente. Dejar el aparato en reposo hasta que la capa líquida superior esté completamente límpida y claramente separada de la capa acuosa. La separación podrá también efectuarse mediante el uso de una centrífuga adecuada (2.6.6). Quitar el tapón y enjuagarlo, así como también el interior del aparato con algunos ml de la mezcla de los disolventes y dejar que los líquidos de los enjuagues penetren en el aparato. Transvasar cuidadosamente al matraz 2.2.3, lo más completamente posible la capa superior por decantación o con ayuda de un sifón (2.6.7). Enjuagar el exterior y el interior del cuello del aparato o el extremo de la parte interior del sifón con unos cuantos mililitros de la mezcla de disolventes. Dejar que los líquidos de los enjuagues de la parte exterior del aparato penetren en el matraz y que los líquidos de los enjuagues de la parte interior del cuello y del sifón penetren en el aparato de extracción. Hacer una segunda extracción repitiendo el procedimiento descrito anteriormente (desde la adición de 25 ml de éter de petróleo), utilizando solamente 15 ml de éter dietílico y 15 ml de éter de petróleo. Hacer una tercera extracción, pero omitiendo el enjuague final.
Evaporar o destilar cuidadosamente la mayor cantidad posible de disolvente (incluido el etanol). Si el matraz es de poca capacidad, parte del disolvente tendrá que eliminarse en la forma citada anteriormente, después de cada extracción. Cuando haya desaparecido el olor a disolvente, calentar el matraz, apoyándolo sobre un lado durante una hora en la estufa. Dejar que el matraz se enfríe a la temperatura ambiente de la balanza y pesar con aproximación de 0,1 mg. Repetir las operaciones de calentar el matraz en estufa y pesar, calentando a intervalos de treinta a sesenta minutos, hasta que se obtenga una masa constante.
Añadir de 15 a 25 ml de éter de petróleo, con objeto de verificar si la materia extraída es totalmente soluble. Calentar ligeramente y agitar el disolvente mediante un movimiento rotatorio, hasta que se haya disuelto toda la grasa. Cuando la materia extraída sea totalmente soluble en el éter de petróleo, la masa de grasa será la diferencia entre las pesadas del matraz 2.2.3 y de la masa constante. En caso contrario extraer completamente la grasa del matraz mediante lavados repetidos con éter de petróleo caliente, dejando que se deposite la materia no disuelta antes de cada decantación. Enjuagar tres veces la parte exterior del cuello del matraz. Calentar el matraz, apoyándolo sobre un lado, durante una hora en la estufa y dejar que se enfríe a la temperatura ambiente de la balanza y pesar con aproximación de 0,1 mg. La masa de grasa será la diferencia entre la masa obtenida anteriormente y esta masa final.
2.4.3 Ensayo en blanco.
Al mismo tiempo que se determina el contenido de grasa de la muestra, efectuar una determinación en blanco con 10 ml de agua destilada, empleando el mismo tipo de aparato de extracción, los mismos reactivos en las mismas cantidades y el mismo procedimiento. Si el resultado del ensayo en blanco excede de 0,5 mg deberán comprobarse los reactivos, y el reactivo o reactivos impuros deberán purificarse o sustituirse.
2.5 Cálculos.
La masa, expresa en gramos, de la muestra extraída es:
(M1 ‒ M2) ‒ (B1 ‒ B2)
y el contenido de grasa en la muestra, expresado en porcentaje, de la masa es:

Siendo:
M1 = masa, en g, del matraz con la materia grasa extraída.
M2 = masa, en g, del matraz sin grasa.
B1 = masa, en g, del matraz del ensayo en blanco después de eliminar los disolventes.
B2 = masa, en g, del matraz del ensayo en blanco.
S = masa, en g, de la porción ensayada.
2.6 Observaciones.
2.6.1 Si no se dispone de etanol se puede utilizar etanol desnaturalizado con metanol, etilmetilcetona, benceno o éter de petróleo.
2.6.2 Para el ensayo de los peróxidos verter 10 ml de éter en una pequeña probeta tapada con tapón de vidrio, previamente enjuagada con éter, añadir 1 ml de solución al 10 por 100 de yoduro de potasio, recién preparada. Agitar y dejar reposar durante un minuto. No debe aparecer ningún color amarillo en ninguna de las capas. El éter dietílico podrá mantenerse exento de peróxidos, añadiendo una lámina de cinc húmeda, que deberá sumergirse completamente en una solución ácida diluida de sulfato de cobre durante un minuto y después lavar con agua. Utilizar por litro una superficie de 80 cm2 aproximadamente de lámina de cinc, cortarla en bandas suficientemente largas para que lleguen por lo menos hasta la mitad del recipiente.
2.6.3 La mezcla de disolventes podrá sustituirse en aquellos casos en que su utilización se haya previsto por éter dietílico o éter de petróleo.
2.6.4 Si la muestra no se pudiera triturar mezclarla cuidadosamente mediante un amasado intenso.
2.6.5 En caso de que haya que retrasar inevitablemente esta operación, tomar todas las precauciones necesarias para asegurar la conservación adecuada de la muestra e impedir la condensación de la humedad en la superficie interior.
2.6.6 Cuando se utilice una centrífuga que no esté provista de un motor trifásico pueden producirse chispas y entonces habrá que tomar las debidas precauciones para evitar explosiones o incendios debido a la presencia de los vapores de éter, por ejemplo, en el caso de una rotura de un tubo.
2.6.7 Si el trasvase no se efectúa mediante un sifón, quizá sea necesario tener que añadir un poco de agua para elevar el plano intermedio entre las dos capas, con objeto de facilitar la decantación.
2.7 Referencia.
1. Código de Principios referente a la Leche y a los Productos Lácteos. Norma B-3. FIL-IDE 5A: 1969.
3. DETERMINACIÓN DEL CONTENIDO DE EXTRACTO SECO
3.1 Principio.
El extracto seco del queso y de los quesos fundidos es la masa, expresada en porcentaje ponderal, que queda después del proceso de desecación.
La precisión del método es de ± 0,1 por 100.
3.2 Material y aparatos.
3.2.1 Balanza analítica, sensibilidad 0,1 mg.
3.2.2 Desecador provisto de un buen deshidratante (gel de sílice con indicador higrométrico o cloruro de calcio).
3.2.3 Estufa de desecación que permita obtener una temperatura constante hasta 110 °C.
3.2.4 Cápsulas de níquel o de aluminio de 2 cm de altura, aproximadamente, y de 6 a 8 cm de diámetro.
3.2.5 Arena de cuarzo de granos gruesos o arena marina purificada con ácido clorhídrico, lavada y calcinada.
3.2.6 Agitadores de vidrio con una extremidad plana.
3.3 Procedimiento.
Colocar 20 g de arena, aproximadamente, y un agitador de vidrio en la cápsula de níquel o de aluminio. Secar la cápsula con la arena y el agitador en la estufa a 105 °C, hasta peso constante. Dejar enfriar la cápsula en el desecador y pesar.
Colocar rápidamente en la cápsula, aproximadamente, 3 g de la muestra de queso preparada y pesar de nuevo.
Triturar cuidadosamente la masa de queso con la arena con ayuda del agitador (3.4.1). Secar la cápsula en la estufa durante cuatro horas a 105 °C. Dejar enfriar en el desecador y pesar.
Proseguir el secado hasta peso constante separando cada pesada por una permanencia en la estufa de media hora.
3.4 Observaciones.
3.4.1 Para los quesos que fundan a la temperatura de 105 °C en una masa córnea, se recomienda guardar primero la cápsula con la masa del queso triturado en el desecador durante dieciséis horas, a la presión atmosférica normal y a la temperatura del laboratorio. Se removerá de vez en cuando el contenido de la cápsula con el agitador, para evitar la formación de costras.
3.5 Referencias.
1. Federación Internacional de Lechería. Norma FIL-IDF 4: 1958.
4. DETERMINACIÓN DEL CONTENIDO EN FÓSFORO
4.1 Principio.
Mineralización de determinada cantidad de muestra con ayuda de ácido sulfúrico en presencia de peróxido de hidrógeno El fosfato se trata con molibdato de sodio y sulfato de hidrazina como agente reductor. El azul de molibdeno así formado se mide por fotometría, calculándose el contenido en fósforo.
La precisión del método es de 0,04 g de fósforo por 100 de producto.
4.2 Material y aparatos.
4.2.1 Balanza analítica
4.2.2 Colorímetro fotoeléctrico que permita lecturas a una longitud de onda de 700 nm.
4.2.3 Aparato apropiado para triturar la muestra.
4.2.4 Matraces Erlenmeyer de 25 ml.
4.2.5 Aparato de mineralización que mantenga los matraces Erlenmeyer en una posición inclinada y provisto de un sistema de calentamiento que no caliente la parte del matraz situada por encima de la superficie del líquido.
4.2.6 Cuerpos que faciliten la ebullición para la mineralización, trozos de porcelana o perlas de vidrio.
4.2.7 Matraces aforados de 50, 100, 200, 500 y 1.000 ml.
4.2.8 Pipetas y/o buretas de 1, 2, 5. 10, 20 y 25 ml.
4.3 Reactivos.
4.3.1 Ácido sulfúrico concentrado (densidad: 1,84 g/ml a 20 °C).
4.3.2 Solución de peróxido de hidrógeno al 30 por 100 (p/v).
4.3.3 Reactivo del molibdato y de sulfato de hidrazina.
4.3.3.1 Solución al 2,5 por 100 de molibdato de sodio.‒Disolver 12,5 g de molibdato de sodio Na2MoO4 • 2H2O en ácido sulfúrico 10 N, completando hasta 500 ml.
4.3.3.2 Solución al 0,15 por 100 de sulfato de hidrazina. Disolver 0,30 g de H2N NH2 • H2SO4, en agua destilada, completando hasta 200 ml.
4.3.3.3 Inmediatamente antes de su empleo, mezclar 25 ml de 4.3.3.1 con 10 ml de 4.3.3.2 y diluir la mezcla hasta 100 ml con agua destilada para preparar el reactivo de molibdato y de sulfato de hidrazina. Esta solución no puede conservarse.
4.3.3.4 Solución normalizada de fosfato.‒Disolver 0,4390 g de fosfato ácido monopotásico KH2P04 en agua destilada hasta obtener una solución de 1.000 ml. Esta solución contiene 100 μg de fósforo en 1 ml. El fosfato de potasio debe haber sido secado durante cuarenta y ocho horas en presencia de un agente de desecación eficaz, por ejemplo, el ácido sulfúrico concentrado. Todos los reactivos deben ser de calidad analítica.
4.4 Procedimiento.
4.4.1 Preparación de la muestra.
Antes del análisis se quitará la corteza o la cara superficial mohosa del queso de modo que se obtenga una muestra representativa del queso tal como se consume habitualmente. La muestra será triturada a continuación en un triturador u otro aparato apropiado y mezclada íntimamente, evitando las pérdidas por evaporación.
La muestra así preparada será conservada en un recipiente al abrigo del aire hasta su análisis, que deberá efectuarse el mismo día.
4.4.2 Determinación.
Introducir sucesivamente en el matraz Erlenmeyer 0,5 g de la muestra, pesados con exactitud de 1 mg, algunas perlas de vidrio o pequeños trozos de porcelana y 4 ml de ácido sulfúrico. Calentar con precaución el matraz Erlenmeyer sobre el aparato de mineralización. Al cesar la formación de espuma, enfriar a la temperatura ambiente; añadir con precaución algunas gotas de la solución de peróxido de hidrógeno, calentar de nuevo y repetir estas operaciones hasta que el contenido del matraz se encuentre límpido e incoloro. Durante el calentamiento, mezclar el contenido del matraz de tiempo en tiempo por agitación. Evitar los recalentamientos locales.
Enjuagar el cuello del matraz con unos 2 ml de agua destilada y calentar de nuevo hasta que el agua se haya evaporado. Dejar hervir el líquido durante una media hora después de la decoloración, con el fin de eliminar todo indicio de peróxido de hidrógeno. Evitar los recalentamientos locales.
Después del enfriamiento a la temperatura ambiente, traspasar el contenido del matraz a un matraz aforado de 100 ml; completar hasta el aforo con agua destilada y mezclar.
Llevar con la pipeta 1 ml de la solución a un matraz aforado de 50 ml y diluir con unos 25 ml de agua destilada. Añadir 20 ml del reactivo de molibdato y de sulfato de hidrazina, llenar el matraz hasta el aforo con agua destilada y mezclar. Colocar el matraz en agua hirviendo y dejar que se forme el color durante quince minutos.
Enfriar a la temperatura ambiente en agua fría y, antes de una hora, medir la densidad óptica con relación al ensayo en blanco (4.4.4) a una longitud de onda de 700 nm.
4.4.3 Preparación de la curva patrón.
Diluir en un matraz aforado 10 ml de la solución normalizada 4 3.4 con agua destilada y completar hasta 100 ml.
Introducir en cinco matraces aforados de 50 ml, 0, 1, 2, 5 y 10 ml de la solución normalizada diluida con el fin de obtener una serie de soluciones testigos que contengan o (valor cero), 10, 20, 50 y 100 μg de fósforo.
Añadir agua destilada a los matraces para obtener un volumen aproximado de 25 ml, añadir 20 ml del reactivo de molibdato y de sulfato de hidrazina, llenar hasta el aforo con agua destilada, mezclar, colocar el matraz con agua hirviendo y dejar que se forme el color durante quince minutos.
Enfriar a la temperatura ambiente en agua fría y medir la densidad óptica de los testigos con respecto al de valor cero, a una longitud de onda de 700 nm.
Determinar la curva patrón señalando las diferencias de densidad óptica con respecto a las cantidades de microgramos de fósforo.
4.4.4 Ensayo en blanco.
Efectuar un ensayo en blanco siguiendo el procedimiento expresado en 4.4.2, pero sin queso.
4.5 Cálculo.
Calcular el contenido en fósforo de la muestra por medio de la fórmula:

Siendo:
P = peso, en μg, de fósforo obtenido al convertir la medida obtenida en el colorímetro utilizando la curva patrón.
W = peso, en g, de la muestra tomada para el análisis.
4.6 Referencia.
1. Federación Internacional de Lechería. Norma FIL-IDF 33A: 1971.
5. DETERMINACIÓN DEL CONTENIDO EN ÁCIDO CÍTRICO
5.1 Principio.
Obtención de un filtrado claro por dispersión de la muestra en agua y clarificación por la adición de ácido tricloroacético. El filtrado se trata con piridina y anhídrido acético y se forma con el ácido cítrico un compuesto de color amarillo. El color obtenido se mide por fotometría.
La precisión del método es de 0,1 g de ácido cítrico anhidro por 100 g de producto.
5.2 Material y aparatos.
5.2.1 Balanza analítica, con precisión que permita de 0,001 g.
5.2.2 Colorímetro fotoeléctrico que permita lecturas a una longitud de onda de 428 nm.
5.2.3 Baño de agua con control termostático que permita una regulación a 32° ± 1 ºC.
5.2.4 Aparato apropiado para triturar la muestra.
5.2.5 Tubos de ensayo con tapones de vidrio o de plástico de 16 ó 18 por 150 mm.
5.2.6 Mortero y mano de porcelana de unos 50 ml.
5.2.7 Matraces aforados de 50, 100 y 1.000 ml.
5.2.8 Pipetas y buretas de 1; 1,3; 4; 5,7; 8; 12; 16, y 20 ml.
5.2.9 Embudos de vidrio de dimensiones apropiadas, por ejemplo, de 5 cm de diámetro.
5.2.10 Papel de filtro duro Whadman número 540, S y S 5893 o equivalente.
5.3 Reactivos.
5.3.1 Ácido tricloroacético al 30 por 100 (p/v).‒Disolver 300 g de ácido tricloroacético en agua destilada y diluir hasta 1.000 ml.
5.3.2 Piridina.
5.3.3 Anhídrico acético
Solución normalizada de citrato.‒Disolver 0,9565 g de citrato trisódico (C6H5O7Na3 • 2H2O) en agua destilada y diluir hasta 1.000 ml.
Todos los reactivos deben ser de calidad analítica.
5.4 Procedimiento.
5.4.1 Preparación de la muestra.
Antes del análisis se quitará la corteza o la capa superficial mohosa del queso de modo que se obtenga una muestra representativa del queso tal como se consume habitualmente. La muestra será triturada a continuación con un triturador u otro aparato apropiado y/o mezclada íntimamente evitando las pérdidas por evaporación. La muestra así preparada será conservada en un recipiente al abrigo del aire hasta su análisis, que deberá efectuarse el mismo día.
5.4.2 Determinación.
Colocar 0,5 g de la muestra, pesada con precisión de 0,001 g en un mortero de porcelana. Dispersar la muestra machacándola con la mano del mortero y añadiendo pequeñas cantidades de agua caliente (60° - 70 °C).
Transpasar el contenido del mortero a un matraz aforado de 100 ml, empleando unos 50 ml de agua. Enfriar a la temperatura ambiente.
Añadir 4. ml de la solución de ácido tricloroacético, mezclar por agitación, llenar con agua destilada hasta el aforo y mezclar de nuevo.
Dejar reposar a la temperatura ambiente durante treinta minutos y filtrar sobre un papel de filtro seco. Desechar la primera porción del filtrado hasta que se obtenga un líquido límpido; se desechará al menos 10 ml.
Introducir con ayuda de una pipeta 1 ml de filtrado claro en un tubo de ensayo provisto de tapón.
Añadir al tubo 1,3 ml de piridina. Mezclar y añadir inmediatamente 5,7 ml de anhídrido acético. Tapar el tubo y mezclar íntimamente su contenido, colocándolo inmediatamente en un baño de agua a 32 °C, dejándolo durante treinta minutos.
Retirar el tubo del baño de María, secarlo y medir la densidad óptica con relación al ensayo en blanco (5.4.4) a una longitud de onda de 428 nm antes de treinta minutos.
5.4.3 Preparación de la curva patrón.
Introducir en seis matraces de 50 ml 0, 4, 8, 12, 16 y 20 ml de la solución normalizada de citrato (5.3.4); añadir a cada matraz agua destilada hasta obtener un volumen aproximado de 25 ml. Añadir 20 ml de la solución de ácido tricloroacético (5.3.1); mezclar por agitación, llenar hasta el aforo con agua destilada y mezclar de nuevo.
Introducir con una pipeta 1 ml de cada solución patrón diluida en tubos de ensayo provistos de tapón, con el fin de obtener una serie de testigos que contengan 0 (valor cero), 50, 100, 150, 200 y 250 μg de ácido cítrico anhidro; añadir a cada tubo 1,3 ml de piridina. Mezclar y añadir inmediatamente 5,7 ml de anhídrido acético. Tapar el tubo y mezclar íntimamente su contenido. Colocar los tubos sin demora en un baño de agua a 32 °C y dejarlos durante treinta minutos.
Retirar los tubos del baño de agua, enfriar a temperatura ambiente, secarlos y medir la densidad óptica de los testigos con relación al valor cero, a una longitud de onda de 428 nm antes de treinta minutos.
Determinar la curva patrón señalando la diferencia de densidad óptica con relación a la cantidad de ácido cítrico anhidro en μg.
5.4.4 Ensayo en blanco.
Efectuar un ensayo en blanco siguiendo el procedimiento expresado anteriormente, pero sin muestra.
5.5 Cálculos.
Calcular el contenido en ácido cítrico anhidro por medio de la fórmula:

Siendo:
C = peso, en μg, de ácido cítrico obtenido al convertir la medida obtenida en el colorímetro utilizando la curva patrón.
W = peso, en g, de la muestra tomada para el análisis.
5.6 Referencia.
1. Federación Internacional de Lechería. Norma FIL-IDF 34B: 1971.
6. DETERMINACIÓN DEL CONTENIDO EN LACTOSA
6.1 Principio.
Preparación de un filtrado claro del queso por dispersión de la muestra en agua y defecación por ferrocianuro de cinc. A una parte del filtrado se le añade una solución que contiene un complejo cúprico. Se determina gravimétricamente el precipitado de óxido cuproso, formado por la acción reductora de la lactosa y los resultados obtenidos se convierten, con la ayuda de tablas, en lactosa anhidra o hidratada.
La precisión del método es de 0,15 g de lactosa anhidra por 100 g de producto.
6.2 Material y aparatos.
6.2.1 Balanza analítica. Sensibilidad, 0,1 mg.
6.2.2 Estufa regulable a 103 °C ± 2 °C.
6.2.3 Mortero de porcelana de un contenido aproximado de 300 ml, de un diámetro interior de unos 110 mm, con una mano apropiada.
6.2.4 Vaso de precipitados de 400 ml.
6.2.5 Crisol filtrante de porcelana de un contenido aproximado de 35 ml y de una porosidad media de 3-15 micras, cuyo peso no debe variar más de 1,0 mg cuando se le aplica el método operatorio descrito más adelante, sin utilizar el queso.
6.2.6 Desecador provisto de un agente de desecación eficaz, como el gel de sílice con indicador de humedad.
6.2.7 Matraz aforado de 500 ml.
6.2.8 Matraz Erlenmeyer de 500 ml.
6.2.9 Pipetas de 25 y 100 ml.
6.2.10 Probeta graduada de 20 ó 25 ml.
6.2.11 Embudo de vidrio de unos 150 mm de diámetro.
6.2.12 Vidrio de reloj destinado a cubrir el vaso de precipitados de 400 ml.
6.2.13 Varilla de vidrio con protección de goma en la punta.
6.2.14 Dispositivo de aspiración de una sección media.
6.2.15 Matraz de vacío con portacrisol.
6.2.16 Filtro plegado o filtro plano de porosidad media, de dimensión correspondiente al embudo 6.2.11.
6.3 Reactivos.
6.3.1 Solución de sulfato de cinc.
Disolver 30 g de sulfato de cinc cristalizado (Zn SO4 • 7H2O) en agua destilada, completando hasta 100 ml.
6.3.2 Solución de ferrocianuro de potasio.
Disolver 15 g de ferrocianuro de potasio cristalizado K4Fe (CN)6 3H2O en agua destilada completando hasta 100 ml.
6.3.3 Solución de sulfato de cobre.
Disolver 70 g de sulfato de cobre (Cu SO4 • 5H2O) en agua destilada, completando hasta 1.000 ml y filtrando si es necesario.
6.3.4 Solución de tartrato alcalino.
Disolver 350 g de tartrato de sodio-potasio (KNa C4H4O5 • 4H2O) y 100 g de hidróxido sódico (NaOH) en agua destilada, completando hasta 1.000 ml. Dejar reposar dos días en un frasco tapado y filtrar. La solución se deteriorará al cabo de un cierto tiempo, lo que puede falsear el ensayo en blanco (resultados más elevados).
6.3.5 Ácido nítrico diluido: HNO3 al 15-20 por 100 en peso.
6.3.6 Alcohol etílico del 96 por 100. Puede ser desnaturalizado con un desnaturalizante apropiado que no deje residuos después de la evaporación.
Los reactivos deben ser de calidad analítica.
6.4 Procedimiento.
6.4.1 Preparación de la muestra para el ensayo.
Antes del análisis se quitará la corteza o capa superficial mohosa del queso, a fin de obtener una muestra representativa del queso tal como habitualmente se consume. La muestra será triturada a continuación en un triturador u otro aparato apropiado y mezclada íntimamente, evitando las pérdidas por evaporación. La muestra así preparada será conservada en un recipiente cerrado hasta su análisis, que deberá efectuarse el mismo día.
6.4.2 Determinación.
Lavar el crisol filtrante con ácido nítrico diluido, enjuagarlo perfectamente con agua caliente y después con 10 ml de alcohol. Secar el crisol a 103 °C ± 2 °C durante treinta minutos, enfriar en un desecador y pesar.
Colocar, aproximadamente, 10 g de la muestra, pesados exactamente, en un mortero de porcelana. Dispersar la muestra machacándola con la mano del mortero, añadiendo pequeñas cantidades de agua caliente (60° - 70 °C). Trasvasar el contenido del mortero a un matraz aforado de 500 ml. Diluir en 400 ml, aproximadamente.
Añadir 5 ml de la solución de sulfato de cinc, mezclando suavemente por rotación del matraz alrededor de su eje, manteniéndolo inclinado. Añadir del mismo modo 5 ml de la solución de ferrocianuro de potasio.
Enfriar el contenido del matraz a 20 °C y completar con agua destilada (a 20 °C) hasta el aforo. Cerrar el matraz con un tapón seco y mezclar íntimamente su contenido mediante una agitación enérgica. Filtrar con un papel de filtro seco, desechando los primeros ml de filtrado.
Tomar con una pipeta 25 ml de la solución de sulfato de cobre (6.3.3) y 25 ml de la solución de tartrato alcalino (6 3.4) y llevarlo a un vaso de precipitado de 400 ml. Mezclar por movimiento de rotación. Calentar la muestre hasta ebullición. Añadir 100 ml del filtrado de la muestra con ayuda de una pipeta. Cubrir el vaso con un vidrio de reloj y calentar de nuevo. Detener el calentamiento exactamente seis minutos después de alcanzar nuevamente el punto de ebullición.
Para asegurar una ebullición más regular y para evitar las proyecciones del líquido, se podrán añadir pequeños trozos de piedra pómez tratados previamente de la misma manera que el crisol y pesados con este último.
Enjuagar el vidrio de reloj con un poco de agua destilada caliente encima del vaso. Trasvasar todo el contenido del vaso a un crisol filtrante preparado previamente (según se indica con anterioridad). Para efectuar este trasvase ayudarse de chorros de agua destilada caliente y de una varilla de vidrio con protección de goma en la punta. El filtrado debe ser de color azul. Si es incoloro, repetir el análisis utilizando una cantidad más pequeña de filtrado diluida en 100 ml.
Enjuagar cuidadosamente el crisol filtrante con agua destilada caliente y después con lo ml de alcohol. Secar el crisol durante treinta minutos a 103 °C ± 2 °C, enfriar en un desecador y pesar.
6.4.3 Ensayo en blanco.
Efectuar un ensayo en blanco siguiendo el procedimiento descrito pero utilizando 10 ml de agua destilada en lugar de 10 g de queso.
6.5 Cálculos.
Corregir la masa de óxido cuproso encontrada en el análisis de la muestra restándole el resultado del ensayo en blanco. Buscar en las tablas la cantidad de lactosa anhidra o hidratada correspondiente a la masa corregida de óxido cuproso.
Calcular el contenido en lactosa anhidra o hidratada de la muestra con ayuda de la fórmula siguiente:

Siendo:
A = masa, en g, de lactosa anhidra o hidratada encontrada en la tabla.
E = masa, en g, de la muestra de ensayo.
V = volumen, en ml, del filtrado utilizado.
0,99 = factor de corrección para compensar el error de volumen que resulta de la presencia de materia grasa y proteínas en la muestra.
6.6 Referencia.
1. Federación Internacional de Lechería. Norma FIL-IDF 43: 1967.
Tabla para determinar la cantidad de lactosa (monohidrataday anhidra) en miligramos, según la cantidad de óxido cuprosoen miligramos
|
Óxido cuproso (Cu2O) en mg |
Lactosa monohidratada (C12H22O11 1 H2O) en mg |
Lactosa anhidra (C12H22O11) en mg |
|---|---|---|
| 10 | 5,1 | 4,8 |
| 11 | 5,8 | 5,5 |
| 12 | 6,4 | 6,1 |
| 13 | 7,1 | 6,7 |
| 14 | 7,7 | 7,3 |
| 15 | 8,4 | 8,0 |
| 16 | 9,0 | 8,6 |
| 17 | 9,7 | 9,2 |
| 18 | 10,3 | 9,8 |
| 19 | 11,0 | 10,5 |
| 20 | 11,6 | 11,0 |
| 21 | 12,3 | 11,7 |
| 22 | 12,9 | 12,3 |
| 23 | 13,6 | 12,9 |
| 24 | 14,2 | 13,5 |
| 25 | 14,8 | 14,1 |
| 26 | 15,5 | 14,7 |
| 27 | 16,2 | 15,4 |
| 28 | 16,8 | 16,0 |
| 29 | 17,7 | 16,6 |
| 30 | 18,1 | 17,2 |
| 31 | 18,7 | 17,8 |
| 32 | 19,4 | 18,4 |
| 33 | 20,0 | 19,0 |
| 34 | 20,7 | 19,7 |
| 35 | 21,3 | 20,2 |
| 36 | 22,0 | 20,9 |
| 37 | 22,6 | 21,5 |
| 38 | 23,3 | 22,1 |
| 39 | 23,9 | 22,7 |
| 40 | 24,6 | 23,4 |
| 41 | 25,2 | 23,9 |
| 42 | 25,9 | 24,6 |
| 43 | 26,5 | 25 2 |
| 44 | 27,2 | 25,8 |
| 45 | 27,8 | 26,4 |
| 46 | 28,5 | 27,1 |
| 47 | 29,1 | 27,6 |
| 48 | 29,8 | 28,3 |
| 49 | 30,4 | 28,9 |
| 50 | 31,4 | 29,5 |
| 51 | 31,7 | 30.1 |
| 52 | 32,4 | 30,8 |
| 53 | 33,0 | 31,4 |
| 54 | 33,7 | 32,0 |
| 55 | 34.3 | 32,6 |
| 56 | 34,9 | 33,2 |
| 57 | 35,3 | 33,8 |
| 58 | 36,2 | 34,4 |
| 59 | 36,9 | 35.1 |
| 60 | 37,5 | 35,6 |
| 61 | 38,2 | 36,3 |
| 62 | 38,8 | 36,9 |
| 63 | 39,4 | 37,4 |
| 64 | 40,1 | 38,1 |
| 65 | 40,8 | 38,8 |
| 66 | 41,4 | 39,3 |
| 67 | 42,0 | 39,9 |
| 68 | 42,7 | 40,6 |
| 69 | 43,3 | 41,1 |
| 70 | 44,0 | 41,8 |
| 71 | 44,6 | 42.4 |
| 72 | 45,3 | 43,0 |
| 73 | 45,9 | 43.6 |
| 74 | 46,6 | 44,3 |
| 75 | 47,2 | 44,8 |
| 76 | 47,9 | 45,5 |
| 77 | 48,5 | 46.1 |
| 78 | 49,2 | 46.7 |
| 79 | 49,8 | 47,3 |
| 80 | 50,4 | 47,9 |
| 81 | 51,1 | 48,5 |
| 82 | 51,8 | 49.2 |
| 83 | 52,4 | 49,8 |
| 84 | 53,1 | 50,4 |
| 85 | 53,7 | 51,0 |
| 86 | 54,4 | 51.7 |
| 87 | 55,0 | 52,3 |
| 88 | 55,7 | 52,9 |
| 89 | 56,3 | 53,5 |
| 90 | 57,0 | 54,3 |
| 91 | 57,6 | 54,7 |
| 92 | 58,2 | 55,3 |
| 93 | 58,9 | 56,0 |
| 94 | 59,5 | 56,5 |
| 95 | 60,2 | 57,2 |
| 96 | 60,8 | 57,8 |
| 97 | 61,4 | 58,3 |
| 98 | 62,1 | 59,0 |
| 99 | 62,8 | 59,7 |
| 100 | 63,4 | 60,2 |
| 101 | 64,0 | 60,8 |
| 102 | 64,6 | 61,4 |
| 103 | 65,3 | 62,0 |
| 104 | 66,0 | 62,7 |
| 105 | 66,6 | 63,3 |
| 106 | 67,2 | 63,8 |
| 107 | 67,9 | 64,5 |
| 108 | 68,6 | 65,2 |
| 109 | 69,2 | 65,7 |
| 110 | 69,9 | 66,4 |
| 111 | 70.5 | 67,0 |
| 112 | 71,2 | 37,6 |
| 113 | 71,9 | 68,3 |
| 114 | 72,5 | 68,9 |
| 115 | 73,2 | 69,5 |
| 116 | 73,8 | 70,1 |
| 117 | 74,5 | 70,8 |
| 118 | 75,1 | 71,3 |
| 119 | 75,8 | 72,0 |
| 120 | 76,5 | 72,7 |
| 121 | 77,1 | 73,2 |
| 122 | 77,7 | 73,8 |
| 123 | 78,4 | 74,5 |
| 124 | 79,1 | 75,1 |
| 125 | 79,8 | 75,8 |
| 126 | 80,4 | 76,4 |
| 127 | 81,0 | 77,0 |
| 128 | 81,7 | 77,6 |
| 129 | 82,3 | 78,2 |
| 130 | 83,0 | 78,9 |
| 131 | 83,7 | 79,5 |
| 132 | 84,4 | 80,2 |
| 133 | 85,0 | 80,8 |
| 134 | 85,6 | 81,3 |
| 135 | 86.3 | 82,0 |
| 136 | 87,0 | 82,7 |
| 137 | 87,7 | 83,3 |
| 138 | 88.3 | 83,9 |
| 139 | 89,0 | 84,6 |
| 140 | 89,6 | 85,1 |
| 141 | 90,3 | 85,8 |
| 142 | 91,0 | 86,5 |
| 143 | 91,6 | 87,0 |
| 144 | 92,2 | 87,6 |
| 145 | 92,9 | 88,3 |
| 146 | 93,6 | 88,9 |
| 147 | 94,3 | 89,6 |
| 148 | 94,9 | 90,2 |
| 149 | 95,6 | 90,8 |
| 150 | 96,2 | 91,4 |
| 151 | 96,9 | 92,1 |
| 152 | 97,6 | 92,7 |
| 153 | 98,2 | 93,3 |
| 154 | 98,8 | 93,9 |
| 155 | 99,5 | 94,5 |
| 156 | 100,2 | 95,2 |
| 157 | 100,8 | 95,8 |
| 158 | 101,5 | 96,4 |
| 159 | 102,2 | 97,1 |
| 160 | 102,8 | 97,7 |
| 161 | 103,5 | 98,3 |
| 162 | 104,2 | 90,0 |
| 163 | 104,9 | 99,7 |
| 164 | 105,6 | 100,3 |
| 165 | 106,2 | 100,9 |
| 166 | 106,9 | 101,6 |
| 167 | 107,6 | 102,2 |
| 168 | 108,2 | 102,8 |
| 169 | 108,9 | 103,5 |
| 170 | 109.6 | 104,4 |
| 171 | 110,2 | 104,7 |
| 172 | 110,9 | 105,4 |
| 173 | 111,6 | 106,0 |
| 174 | 112,3 | 106,7 |
| 175 | 113,0 | 107,4 |
| 176 | 113,6 | 107,9 |
| 177 | 114,3 | 108,6 |
| 178 | 115,0 | 109,3 |
| 179 | 115,6 | 109,8 |
| 180 | 116,3 | 110,5 |
| 181 | 117,0 | 111,2 |
| 182 | 117,6 | 111,7 |
| 183 | 118,3 | 112,4 |
| 184 | 119,0 | 113,1 |
| 185 | 119,7 | 113,7 |
| 186 | 120.3 | 114,3 |
| 187 | 121,0 | 115,0 |
| 188 | 121,7 | 115,6 |
| 189 | 122,4 | 116,3 |
| 190 | 123,0 | 116,9 |
| 191 | 123,7 | 117,5 |
| 192 | 124,3 | 118,1 |
| 193 | 125,0 | 118,8 |
| 194 | 125,6 | 119,3 |
| 195 | 126,3 | 120,0 |
| 196 | 127,0 | 120,7 |
| 197 | 127,7 | 121,3 |
| 198 | 128,4 | 122,0 |
| 199 | 129,1 | 122,6 |
| 200 | 129,7 | 123,2 |
| 201 | 130,4 | 123,9 |
| 202 | 131,1 | 124,5 |
| 203 | 131,8 | 125,2 |
| 204 | 132,4 | 125,8 |
| 205 | 133,1 | 126,4 |
| 206 | 133,8 | 127,1 |
| 207 | 134,5 | 127,8 |
| 208 | 135,2 | 128,4 |
| 209 | 135,8 | 129,0 |
| 210 | 136,5 | 129,7 |
| 211 | 137,2 | 130,3 |
| 212 | 137,9 | 131,0 |
| 213 | 138,6 | 131,7 |
| 214 | 139,3 | 132,3 |
| 215 | 140,0 | 133,0 |
| 216 | 140,6 | 133,6 |
| 217 | 141,3 | 134,2 |
| 218 | 142,0 | 134,9 |
| 219 | 142.6 | 135.5 |
| 220 | 143,3 | 136,1 |
| 221 | 144,0 | 136,8 |
| 222 | 144.7 | 137.5 |
| 223 | 145,4 | 138,1 |
| 224 | 146,1 | 138,7 |
| 225 | 146,8 | 139,5 |
| 226 | 147,5 | 140,1 |
| 227 | 148,1 | 140,7 |
| 228 | 148,8 | 141,4 |
| 229 | 149,4 | 141,9 |
| 230 | 150,1 | 142,6 |
| 231 | 150,8 | 143,3 |
| 232 | 151,4 | 143,8 |
| 233 | 152,1 | 144,5 |
| 234 | 152,8 | 145,2 |
| 235 | 153,4 | 145,7 |
| 236 | 154,1 | 146,4 |
| 237 | 154,8 | 147,1 |
| 238 | 155,4 | 147,6 |
| 239 | 156,1 | 148,3 |
| 240 | 156,9 | 149,1 |
| 241 | 157,4 | 149,5 |
| 242 | 158,1 | 150,2 |
| 243 | 158,7 | 150,8 |
| 244 | 159,4 | 151,4 |
| 245 | 160,1 | 152,1 |
| 246 | 160,7 | 152,7 |
| 247 | 161,4 | 153,3 |
| 248 | 162,0 | 153,9 |
| 249 | 162,7 | 154,6 |
| 250 | 163,4 | 155,2 |
| 251 | 164,0 | 155,8 |
| 252 | 164.7 | 156.5 |
| 253 | 165,4 | 157,4 |
| 254 | 166,0 | 157,7 |
| 255 | 166.7 | 158.4 |
| 256 | 167,3 | 158,9 |
| 257 | 168,0 | 159.6 |
| 258 | 168.7 | 160,3 |
| 259 | 169,4 | 160,9 |
| 260 | 170,0 | 161,5 |
| 261 | 170,7 | 162,2 |
| 262 | 171,3 | 162,7 |
| 263 | 172,0 | 163,4 |
| 264 | 172,6 | 164,0 |
| 265 | 173,3 | 164,6 |
| 266 | 174,0 | 165,3 |
| 267 | 174,7 | 166,0 |
| 268 | 175,4 | 166,6 |
| 269 | 176,1 | 167,3 |
| 270 | 176,8 | 168,0 |
| 271 | 177,5 | 168,6 |
| 272 | 178,2 | 169,3 |
| 273 | 178,8 | 169,9 |
| 274 | 179,5 | 170,5 |
| 275 | 180,2 | 171,2 |
| 276 | 180,9 | 171,9 |
| 277 | 181,6 | 172,5 |
| 278 | 182,3 | 173,2 |
| 279 | 183,0 | 173,9 |
| 280 | 183,6 | 174,4 |
| 281 | 184,3 | 175,1 |
| 282 | 185,0 | 175,8 |
| 283 | 185,7 | 176,4 |
| 284 | 186,4 | 177,1 |
| 285 | 187,1 | 177,7 |
| 286 | 187,8 | 178,4 |
| 287 | 188,5 | 179,1 |
| 288 | 189,1 | 179,6 |
| 289 | 189,8 | 180,3 |
| 290 | 190,5 | 181,0 |
| 291 | 191,2 | 181,6 |
| 292 | 191,9 | 182.3 |
| 293 | 192,6 | 183,0 |
| 294 | 193,3 | 183,6 |
| 295 | 194,0 | 184.3 |
| 296 | 194,7 | 185,0 |
| 297 | 195,4 | 185,6 |
| 298 | 196,0 | 186,2 |
| 299 | 196,7 | 186,9 |
| 300 | 197,4 | 187,5 |
| 301 | 198,1 | 188,2 |
| 302 | 198,8 | 188,9 |
| 303 | 199,5 | 189,5 |
| 304 | 200,2 | 190,2 |
| 305 | 200,9 | 190,9 |
| 306 | 201,6 | 191,5 |
| 307 | 202,3 | 192,2 |
| 308 | 203,0 | 192,9 |
| 309 | 203,7 | 193,5 |
| 310 | 204,4 | 194,2 |
| 311 | 205,2 | 194,9 |
| 312 | 205,9 | 195,6 |
| 313 | 206,6 | 196,3 |
| 314 | 207,3 | 196,9 |
| 315 | 208,0 | 197,6 |
| 316 | 208,7 | 198,3 |
| 317 | 209,5 | 199,0 |
| 318 | 210,2 | 199,7 |
| 319 | 210,9 | 200,4 |
| 320 | 211,6 | 201,0 |
| 321 | 212,3 | 201,7 |
| 322 | 213,0 | 202,4 |
| 323 | 213,7 | 203,0 |
| 324 | 214,4 | 203,7 |
| 325 | 215,2 | 204,4 |
| 326 | 215,9 | 205,1 |
| 327 | 216,6 | 205,8 |
| 328 | 217,3 | 206,4 |
| 329 | 218,0 | 207,1 |
| 330 | 218,8 | 207,9 |
| 331 | 219,5 | 208,5 |
| 332 | 220,2 | 209,2 |
| 333 | 220,9 | 209,9 |
| 334 | 221,6 | 210,5 |
| 335 | 222,4 | 211,3 |
| 336 | 223,1 | 211,9 |
| 337 | 223,8 | 212,6 |
| 338 | 224,5 | 213,3 |
| 339 | 225,2 | 213,9 |
| 340 | 225,9 | 214,6 |
| 341 | 226,6 | 215,3 |
| 342 | 227,2 | 215,8 |
| 343 | 227,9 | 216,5 |
| 344 | 228,6 | 217,2 |
| 345 | 229,3 | 217,8 |
| 346 | 230,0 | 218,5 |
| 347 | 230,7 | 219,2 |
| 348 | 231,4 | 219,8 |
| 349 | 232,1 | 220,5 |
| 350 | 232,8 | 221,2 |
| 351 | 233,5 | 221,8 |
| 352 | 234,2 | 222,5 |
| 353 | 234,9 | 223,2 |
| 354 | 235,6 | 223,8 |
| 355 | 236,2 | 224,4 |
| 356 | 237,0 | 225,2 |
| 357 | 237,7 | 225,8 |
| 358 | 238,4 | 226,5 |
| 359 | 239,1 | 227,1 |
| 360 | 239,8 | 227,8 |
| 361 | 240,5 | 228,5 |
| 362 | 241,2 | 229,1 |
| 363 | 241,8 | 229,7 |
| 364 | 242,5 | 230,4 |
| 365 | 243,2 | 231,0 |
| 366 | 243,9 | 231,7 |
| 367 | 244,6 | 232,4 |
| 368 | 245,2 | 232,9 |
| 369 | 245,9 | 233,6 |
| 370 | 246,6 | 234,3 |
| 371 | 247,3 | 234,9 |
| 372 | 248,0 | 235,6 |
| 373 | 248,7 | 236.3 |
| 374 | 249,4 | 236,9 |
| 375 | 250,1 | 237,6 |
| 376 | 250,8 | 238.3 |
| 377 | 251,3 | 239,0 |
| 378 | 252,3 | 239,7 |
| 379 | 253,0 | 2^0,4 |
| 380 | 253,7 | 241,0 |
| 381 | 254,4 | 241,7 |
| 382 | 255,1 | 242,3 |
| 383 | 255,8 | 243,0 |
| 384 | 256,6 | 243,8 |
| 385 | 257,3 | 244,4 |
| 386 | 258,0 | 245,1 |
| 387 | 258,7 | 245,8 |
| 388 | 259,5 | 246,5 |
| 389 | 260,2 | 247,2 |
| 390 | 260,9 | 247,9 |
| 391 | 261,6 | 248,5 |
| 392 | 262,3 | 249,2 |
| 393 | 263,1 | 249,9 |
| 394 | 263,8 | 250,6 |
| 395 | 264,5 | 251,3 |
| 396 | 265,2 | 251,9 |
| 397 | 265,9 | 252,6 |
| 398 | 266,7 | 253,4 |
| 399 | 267,4 | 254,0 |
| 400 | 268,1 | 254,7 |
| 401 | 268,8 | 255,4 |
| 402 | 269,6 | 256,1 |
| 403 | 270,3 | 256,8 |
| 404 | 271,0 | 257,5 |
| 405 | 271,8 | 258,2 |
| 406 | 272,5 | 258,9 |
| 407 | 273,2 | 259,5 |
| 408 | 274,0 | 260.3 |
| 409 | 274,7 | 261,0 |
| 410 | 275,5 | 261,7 |
| 411 | 276,2 | 262.4 |
| 412 | 276,9 | 263,1 |
| 413 | 277,7 | 263,8 |
| 414 | 278,4 | 264,5 |
| 415 | 279,1 | 265,1 |
| 416 | 279,9 | 265,9 |
| 417 | 280,6 | 266,6 |
| 418 | 281,4 | 267,3 |
| 419 | 282,2 | 268,1 |
| 420 | 283,0 | 268,9 |
| 421 | 283,7 | 269,5 |
| 422 | 234,5 | 270,3 |
| 423 | 285,2 | 270,9 |
| 424 | 286,0 | 271,7 |
| 425 | 286,8 | 272,5 |
| 426 | 287,6 | 273,2 |
| 427 | 288,3 | 273,9 |
| 428 | 289,1 | 274.6 |
| 429 | 289,9 | 275,4 |
| 430 | 290,7 | 276,2 |
| 431 | 291,4 | 276,8 |
| 432 | 292,2 | 277,6 |
| 433 | 293,0 | 278,4 |
| 434 | 293,8 | 279,1 |
| 435 | 294,5 | 279,8 |
| 436 | 295,3 | 280,5 |
| 437 | 296,0 | 281,2 |
| 438 | 296,8 | 282,0 |
| 439 | 297,6 | 282,7 |
| 440 | 298,4 | 283,5 |
| 441 | 299,2 | 284,2 |
| 442 | 299,9 | 284,9 |
| 443 | 300,7 | 285,7 |
| 444 | 301,4 | 286,3 |
| 445 | 302,2 | 287,1 |
| 446 | 303,0 | 287,9 |
| 447 | 303,7 | 288,5 |
| 448 | 304,5 | 289,3 |
| 449 | 305,2 | 289,9 |
| 450 | 306,0 | 290,7 |
LECHE
4. LACTOSA
4.1 Principio.
Después de desproteinización y filtración de la leche, la lactosa se determina por valoración de la cantidad de halógeno reducido en el curso de la reacción entre la lactosa y el conjunto ioduro de potasio cloramina T.
Este método es aplicable a la leche natural, no alterada, así como a las conservadas por adición de formaldehido (1:2.500).
El procedimiento es el que figura en el método número 4 del anejo III de la Orden de 31 de enero de 1977 («Boletín Oficial del Estado» de 14 de julio).
9. SACAROSA
(Determinación polarimétrica en la leche condensada)
9.1 Principio.
La determinación se basa en el principio de inversión de Clerget: mediante un tratamiento suave por ácido se hidroliza completamente la sacarosa, mientras la lactosa y los otros azúcares prácticamente no se hidrolizan. El contenido en sacarosa se deduce del cambio del poder rotatorio de la solución y se expresa en porcentaje en peso.
Este método es aplicable a leche condensada, entera o desnatada.
El procedimiento es el que figura en el método número 9 del anejo III de la Orden de 31 de enero de 1977 («Boletín Oficial del Estado» de 14 de julio).
14(a). HARINA DE ALFALFA
(Método comparativo)
14(a).1 Principio.
Determinación de harina de alfalfa en leche desnaturalizada por comparación visual y microscópica frente a muestras patrón.
14(a).2 Material y aparatos.
14(a).2.1 Tamiz de 0,25 mm de luz de malla.
14(a).2.2 Lupa para 10 a 20 aumentos.
14(a).2.3 Material necesario para observación microscópica.
14(a).3 Procedimiento.
14(a).3.1. Preparación de la muestra patrón de harina de alfalfa.
Mezclar íntimamente muestras de harina de alfalfa adquiridas en el mercado, finamente molidas.
Tamizar la harina obtenida empleando el tamiz 14(a).2.1.
Separar las dos fracciones y mezclarlas en la siguiente proporción: 98 partes de la fracción más fina y 2 de la más gruesa.
Dicha mezcla se considera como muestra patrón de harina de alfalfa.
14(a).3.2 Preparación de las muestras patrón de leche en polvo-harina de alfalfa.
Mezclar y homogeneizar leche en polvo sin desnaturalizar y la muestra patrón de harina de alfalfa obtenida, según las siguientes proporciones:
| Gramos de leche en polvo. | 99 | 98,5 | 98 | 97,5 | 97 | 96,5 | 96 |
| Gramos de harina de alfalfa. | 1 | 1,5 | 2 | 2,5 | 3 | 3,5 | 4 |
| Porcentaje de harina de alfalfa. | 1 % | 1,5 % | 2 % | 2,5 % | 3 % | 3,5 % | 4 % |
Realizar con estas muestras patrón una serie de preparaciones, colocando en el centro de un portaobjetos 0,6 g aproximadamente de cada una de las muestras. Cubrir con otro portaobjetos y extender el polvo mediante compresión y giro de los dos vidrios, hasta lograr una lámina circular de unos 2 cm de diámetro. Sujetar los dos portaobjetos fijando los extremos con papel adhesivo transparente.
La colección de patrones así obtenida se conserva para ulteriores determinaciones.
14(a).3.3 Determinación de la harina de alfalfa contenida en la muestra de leche en polvo a analizar.
Obtener una preparación en forma análoga a la anteriormente descrita y comparar con las muestras patrón de leche en polvo-harina de alfalfa, separando aquellas que, a simple vista, sean análogas o muy próximas a la muestra problema.
Proceder a una comparación microscópica entre la muestra a analizar y cada una de las muestras separadas, empleando lupa de 10 a 20 aumentos, considerando tanto el tamaño como la densidad de las partículas que aparecen en una serie de campos, obteniendo valores medios y expresando los resultados en porcentaje de harina de alfalfa contenida en la leche en polvo analizada.
14(a).4 Referencias.
1. A López, M del C. Díaz-Peñalver y J. Gutiérrez: «Leches desnaturalizadas. Comprobación de la desnaturalización de las mismas».
14(b). HARINA DE ALFALFA
(Método gravimétrico)
14(b).1 Principio.
Separación de las partículas de alfalfa por lavado con agua y posterior determinación gravimétrica.
14(b).2 Material y aparatos.
14(b).2.1 Vaso de precipitados de 250 ml.
14(b).2.2 Tamiz de 0,25 mm de luz de malla.
14(b).2.3 Embudo.
14(b).2.4 Matraz Erlenmeyer de 2.000 ml.
14(b).2.5 Trompa de vacío.
14(b).2.6 Placa filtrante de vidrio del número 1.
14(b).2.7 Estufa de desecación.
14(b).3 Procedimiento.
Pesar 50 g de leche desnaturalizada en un vaso de precipitados de 250 ml y adicionar 150 ml de agua. Una vez bien impregnada la leche desnaturalizada, formar una papilla fina por agitación mediante una varilla con el extremo curvado en forma de U.
Añadir a continuación otros 100 ml de agua, agitar y pasar la papilla a través del tamiz 14(b).2.2 (previamente desecado a 100 °C hasta peso constante, con precisión de 0,01 g) colocado sobre un embudo y éste a su vez sobre un matraz Erlenmeyer de 2.000 ml (fig. 1).
Lavar el vaso y el tamiz añadiendo poco a poco agua a 40 °C en chorro fino, removiendo los pequeños grumos con ayuda de la varilla hasta que toda la leche haya sido arrastrada por el agua. Se precisan aproximadamente 1,5 litros.
Dejar escurrir el tamiz y desecarlo en estufa a 100 °C, colocado sobre un papel de filtro.
Una vez desecado el tamiz hasta peso constante, se pesa con aproximación de 0,0i g, recogiéndose en él previamente las partículas que pudieran haber caído en el papel de filtro a medida que se producía la desecación de la harina de alfalfa. Se obtiene así un peso p1.
Las partículas de alfalfa, a causa de la humedad, sufren un hinchamiento, por lo que es necesario tamizar de nuevo después de la desecación y de exponer el tamiz a humedad ambiente durante dos horas, obteniéndose así el peso p2 de la cantidad retenida por el mismo.
Filtrar a vacío la leche recogida en el Erlenmeyer a través de la placa filtrante 14(b).2.6, cuyo fondo y paredes se han recubierto de algodón (fig. 2). Antes de iniciarse la filtración, pesar con aproximación de 0,01 g, la placa y el algodón previamente desecados a 100 °C. La filtración se realiza añadiendo la leche poco a poco sobre el centro de la placa, cuidando que el vacío no sea demasiado intenso para evitar la formación de espuma. Lavar con abundante agua a 40 °C hasta que ésta pase completamente transparente.
Desecar la placa a 100 °C hasta peso constante y pesar con aproximación de 0,01 g. Se obtiene así el peso de las partículas de alfalfa, P3.
14(b).4 Cálculos.
La humedad propia de la harina de alfalfa se estima en un 8 por 100. Los valores reales de la harina de alfalfa total contenida en 50 gramos de muestra (P) y de la retenida por el tamiz (p), serán:


14(b).5 Referencias.
1. A. López, M. del C. Díaz-Peñalver y J. Gutiérrez: «Leches desnaturalizadas. Comprobación de la desnaturalización de las mismas».
15(a). FENOLFTALEÍNA EN LECHE DESNATURALIZADA
(Método cualitativo)
15(a).1 Principio.
Detección de fenolftaleína en leche desnaturalizada en presencia de una solución alcalina.
15(a).2 Material y aparatos.
Tubos de ensayo 160/60.
15(a).3 Reactivos.
Solución de hidróxido sódico 2 N.
15(a).4 Procedimiento.
Poner en un tubo de ensayo 160/60, aproximadamente, 0,5 g de leche en polvo, añadir 10 ml de agua destilada y agitar hasta lograr una emulsión uniforme. Añadir 2 ml de solución 2 N de NaOH.
La presencia de fenolftaleína en la muestra se manifiesta por la aparición de una serie de puntos rojo-rosados que se ensanchan, intensificando su color hasta llegar a un máximo, para empalidecer seguidamente y llegar a desaparecer transcurrido un cierto tiempo.
Comparar con un patrón de leche en polvo conteniendo fenolftaleína al 1/20.000.
15(b). FENOLFTALEÍNA EN YOGUR, GALLETAS Y CHOCOLATE
(Método cualitativo)
15(b).1 Principio.
Extracción y concentración de la fenolftaleína presente en el alimento e identificación por viraje a color rojo en medio alcalino que desaparece en medio ácido. La separación y concentración de la fenolftaleína se realiza por extracción etérea y soluciones alcalinas.
15(b).2 Material y aparatos.
15(b).2.1 Centrífuga que alcance 3.000 r.p.m.
15(b).2.2 Aparato de destilación.
15(b).3 Reactivos.
15(b).3.1 Éter etílico.
15(b).3.2 Solución de hidróxido sódico al 1 por 100.
15(b).3.3 Ácido sulfúrico al 5 por 100.
15(b).4 Procedimiento.
Pesar 50 g de la muestra, reducida a polvo, si es necesario, y llevar a un matraz Erlenmeyer de 250 ml de capacidad. Añadir 50 ml de éter etílico. Agitar suavemente durante 3 a 5 minutos.
Centrifugar la mezcla así obtenida a 1.500-2.000 r.p.m. durante diez minutos. Recoger el líquido sobrenadante.
Volver a emulsionar el sedimento con nueva aportación de éter etílico, agitando durante dos-tres minutos.
Centrifugar en las mismas condiciones y recoger el sobrenadante añadiéndolo a la centrifugación anterior.
Repetir esta operación por tercera vez. Reunir todas las porciones de extracto etéreo y destilar para eliminar el éter, tomando las precauciones habituales en este tipo de destilación. Queda un residuo graso abundante.
Añadir al residuo graso solución acuosa de hidróxido sódico al 1 por 100. Agitar suavemente durante 2-3 minutos. Si es necesario, calentando suavemente.
Centrifugar la emulsión obtenida a 2.000-3.000 r.p.m. durante cinco minutos.
Eliminar la capa superior grasa y tomar la fracción acuosa inferior. Cuando el producto contenga fenolftaleína, la capa acuosa presenta una coloración roja-rosada que desaparece al acidificar con ácido sulfúrico al 5 por 100 y vuelve a reaparecer si se alcaliniza el medio.
16. FÉCULA
(Método comparativo)
16.1 Principio.
Determinación de fécula en leche desnaturalizada por comparación con muestras patrón.
16.2 Material y aparatos.
16.2.1 Matraz aforado de 100 ml.
16.2.2 Probeta de 100 ml.
16.2.3 Pipeta de 10 ml.
16.3 Reactivos.
Solución de iodo 0,1 N.
16.4 Procedimiento.
16.4.1 Preparación del desnaturalizante patrón.
Mezclar íntimamente 80 g de salvado y 20 g de fécula de la misma naturaleza que la que se pretende identificar.
16.4.2 Preparación de patrones de leche descremada en polvo y desnaturalizante patrón.
Mezclar haciendo uso del mortero, según las siguientes proporciones:
| Patrón |
Leche descremada en polvo (g) |
Desnaturalizante patrón (g) |
|---|---|---|
| Núm. 1. | 85 | 15 |
| Núm. 2. | 80 | 20 |
| Núm. 3. | 75 | 25 |
16.4.3 Determinación de la muestra.
Pesar exactamente un gramo de la muestra con precisión de 0,01 gramos. Pasar a un mortero y preparar una papilla con 25 ml de agua hirviente y verter en un matraz aforado de 100 ml. Lavar el mortero con agua caliente repetidas veces vertiendo los líquidos de lavado en el matraz hasta obtener unos 75 ml, agitar enérgicamente y llevar a un baño de María durante diez minutos, agitando de cuando en cuando; enfriar el matraz al chorro de agua fría y llevar el contenido exactamente a 100 ml.
Tomar 10 ml exactamente medidos y pasar a una probeta de 100 ml. Añadir 80 ml de agua destilada, 1 ml de solución I2 0,1 N, completar hasta el enrase con agua destilada: agitar enérgicamente.
Verificar el mismo método con los tres patrones preparados. El color desarrollado en la muestra se compara a los cinco minutos con el de los tres patrones.
17. SALVADO
17.1 Principio.
Separación por tamizado del salvado y determinación gravimétrica.
17.2 Material y aparatos.
17.2.1 Tamiz de 0,210 mm de luz de malla.
17.2.2 Tamiz de 0,074 mm de luz de malla.
17.2.3 Estufa de desecación.
17.3 Procedimiento.
Pesar con precisión de 0,01 g, 20 g de leche y tamizarla exhaustivamente a través del tamiz 17.2.1 estándar. Recoger cuidadosamente la fracción de salvado retenida por el tamiz y pesar
A continuación, empastar con agua la parte tamizada, deshaciendo todos los grumos que puedan formarse con ayuda de una varilla de vidrio con el extremo curvado en forma de U y diluir con agua hasta completar unos 300 ml.
Verter la emulsión sobre el tamiz 17.2.2, previamente desecado a 100 °C hasta peso constante; lavar repetidamente el residuo que permanece en el tamiz hasta que las aguas de lavado pasen transparentes e incoloras. Desecar el tamiz en una estufa a 100 ºC hasta peso constante.
17.4 Cálculos.
El contenido en salvado en 100 gramos de la muestra viene dado por:
Ps = 5 (a + 1,3 b)
Siendo:
b = peso, en g, de la fracción de salvado retenida por el tamiz de 17.2.1.
a = peso, en g, de la fracción de salvado retenida por el tamiz de 17.2.1,
17.5 Observaciones.
17.5.1 Se estima que la pérdida que sufre el salvado por lavado y desecación es del 30 por 100.
18. FÉCULA + SALVADO
18.1 Principio.
Separación por centrifugación de la fécula y del salvado y determinación gravimétrica.
18.2 Material y aparatos.
18.2.1 Centrífuga de 3.000 r.p.m.
18.2.2 Estufa de desecación al vacío.
18.3 Reactivos.
Solución yodo-yodurada. Mezclar 1 g de iodo y 2 g de ioduro potásico en agua destilada hasta 200 ml. Mantener la solución en el frasco cuentagotas.
18.4 Procedimiento.
Pesar con aproximación de 0,001 g, 0,5 g de la muestra de leche a analizar y colocarla en un tubo de centrífuga previamente pesado con igual exactitud. Añadir 10 ml de agua destilada fría y agitar con una varilla de vidrio hasta disolución de la leche. A continuación, centrifugar a 3.000 r.p.m.
Decantar la emulsión sobrenadante y añadir 10 ml de agua destilada fría al residuo insoluble; agitar de nuevo con una varilla, centrifugar otros quince minutos a idénticas revoluciones y decantar. Repetir estas operaciones al menos cuatro veces.
Los líquidos obtenidos por decantación tras la centrifugación no deben dar coloración azul con solución yodo-yodurada.
Desecar a 80 °C en estufa de vacío el residuo final contenido en el tubo de centrífuga, hasta peso constante. El peso del residuo representará el peso total del desnaturalizante contenido en 0,5 g de la leche desnaturalizada.
18.5 Cálculo.
El salvado contiene una humedad media que puede cifrarse en un 10 por 100; por ello, el peso del residuo centrifugado debe incrementarse en la humedad correspondiente al salvado contenido en 0,5 g de leche, que en un producto correctamente desnaturalizado al 20 por 100 será 0,008 g.
El peso del desnaturalizante total será:

Siendo:
Rs = peso, en g, del residuo.
h = factor de corrección de humedad del salvado = 0,008 g.
P = peso, en g, de la leche desnaturalizada.
1. UREA
1.1 Principio.
La urea se determina gravimétricamente por precipitación con xanthidrol. Este método es aplicable a productos lácteos.
1.2 Material y aparatos.
1.2.1 Matraz aforado de 100 ml de capacidad.
1.2.2 Baño de agua.
1.2.3 Crisol de fondo poroso.
1.2.4 Estufa de desecación, regulable a 105 °C.
1.2.5 Balanza de precisión.
1.3 Reactivos.
1.3.1 Alcohol de 96°.
1.3.2 Ácido acético glacial.
1.3.3 Solución de xanthidrol.
1.3.4 Alcohol de 96° saturado de dixantil-urea.
Añadir por cada gramo de urea 6,60 g de xanthidrol, es decir, 66 ml de xanthidrol al 10 por 100 en metanol, formándose un precipitado de dixantil-urea. Filtrar por crisol filtrante, quedando retenido el precipitado. Pasar por el mismo crisol alcohol de 96° con lo que se obtiene el alcohol de 96° saturado de dixantil-urea.
1.4 Procedimiento.
Pesar unos 3 g del producto e introducirlo en un matraz aforado de 100 ml que contenga 20 ml de agua destilada y agitar durante una hora. Enrasar con alcohol de 96° y agitar de nuevo durante unos quince minutos. Dejar reposar unas dos horas y filtrar por papel de filtro.
Poner 50 ml de filtrado en un vaso (25 ml si se trata de un producto rico en urea) y concentrar al baño de agua hirviendo hasta un volumen de unos 10 ml. Añadir 35 ml de ácido acético glacial y 2 ml de solución recientemente preparada de xanthidrol al 10 por 100 de metanol, repitiendo la adición otras cuatro veces a intervalos de cinco a diez minutos. Dejar reposar durante una hora, como mínimo, y filtrar el precipitado formado a través de un criso1 de fondo poroso previamente tarado.
Lavar tres veces empleando cada vez 5 ml de alcohol de 96° saturado de dixantil-urea, desecar en estufa a 105 °C hasta peso constante y pesar.
1.5 Cálculos.
1 g de dixantil-urea = 0,143 g de urea.

Siendo:
P = peso, en g, de dixantil-urea encontrado.
P’ = peso, en g, de muestra analizada.
2. ALCALOIDES EN ALTRAMUCES
2.1 Principio.
Los alcaloides son puestos en solución en una mezcla de éter dietílico y de cloroformo, extrayéndose por ácido clorhídrico. Los alcaloides son precipitados por el ácido sílico-túngstico incinerados y pesados.
2.2 Material y aparatos.
2.2.1 Agitador mecánico.
2.2.2 Cápsula de incineración de platino, cuarzo o porcelana.
2.2.3 Horno de mufla eléctrico.
2.3 Reactivos.
2.3.1 Éter dietílico.
2.3.2 Cloroformo.
2.3.3 Solución de hidróxido sódico 4 N.
2.3.4 Ácido clorhídrico 0,3 N.
2.3.5 Cloruro sódico.
2.3.6 Solución al 10 por 100 (p/v) de ácido sílico-túngstico SiO212WO3 • 26H2O.
2.4 Procedimiento.
Pesar, con aproximación de 5 mg, 15 g de la muestra e introducirla en un recipiente de unos 200 ml provisto de tapón esmerilado. Añadir 100 ml de éter dietílico y 50 ml de cloroformo exactamente medidos e inmediatamente y con la ayuda de una bureta graduada, 10 ml de la solución de hidróxido sódico. Agitar vigorosamente al principio para evitar la formación de grumos, continuando la agitación a intervalos; dejar reposar hasta el día siguiente. Si el líquido sobrenadante no está totalmente limpio, añadir algunas gotas de agua; filtrar la capa de éter-cloroformo. Recoger 50 ml de filtrado en un matraz aforado de 50 ml y traspasarlos cuantitativamente con la ayuda de 50 ml de éter dietílico a una ampolla de decantación de 150 ml. Extraer tres veces sucesivas con 20 ml de ácido clorhídrico, dejar decantar y recoger el extracto ácido después de cada extracción. Reunir los extractos ácidos en un matraz de 250 ml y eliminar las últimas trazas de éter y del cloroformo calentando ligeramente. Añadir alrededor de 1 g de cloruro de sodio, dejar enfriar y precipitar los alcaloides mediante la solución del ácido sílico-túngstico (2.6.1). Agitar mecánicamente durante treinta minutos, dejar reposar durante una noche, filtrar sobre filtro de cenizas conocidas y lavar el precipitado sucesivamente por dos veces con 10 ml y dos veces con 5 ml de ácido clorhídrico.
Situar el filtro que contiene el precipitado en una cápsula de incineración e incinerar a 900 °C. Dejar enfriar y pesar.
2.5 Cálculos.
% alcaloides = 0,2 P
Siendo:
P = peso de las cenizas.
Expresar los resultados en porcentaje de la muestra.
2.6 Observaciones.
2.6.1 Añadir solución sílico-túngstica hasta que se vea que no se forma más precipitado blanco lechoso de alcaloide.
2.7 Referencias.
1. Journal Officiel des Communautés Européennes. Número 155/36.
3. PROTEÍNA TOTAL
(Proteína bruta)
3.1 Principio.
Mineralizar la muestra por vía húmeda y alcalinizar por medio de solución de hidróxido sódico. El amoníaco liberado es arrastrado por destilación y recogido en una cantidad determinada de ácido sulfúrico, valorando el exceso con solución de hidróxido sódico.
3.2 Material y aparatos.
3.2.1 Mineralizador y destilador Kjeldahl.
3.3 Reactivos.
3.3.1 Sulfato de potasio.
3.3.2 Catalizador.‒Óxido de cobre (CuO) o sulfato de cobre cristalizado (SO4Cu • 5H2O).
3.3.3 Cinc granulado.
3.3.4 Ácido sulfúrico d = 1,84.
3.3.5 Ácido sulfúrico 0,1 N.
3.3.6 Ácido sulfúrico 0,5 N.
3.3.7 Indicador de fenolftaleína.‒Disolver 100 mg de fenolftaleína en 100 ml de etanol del 70 por 100 (v/v).
3.3.8 Disolver 300 mg de rojo de metilo en 100 ml de etanol del 95-96 por 100 (v/v).
3.3.9 Solución de hidróxido sódico al 30 por 100 (p/v).
3.3.10 Solución de hidróxido sódico 0,1 N.
3.3.11 Solución de hidróxido sódico 0,25 N.
3.3.12 Solución saturada de sulfuro sódico.
3.3.13 Solución de sulfuro potásico al 4 por 100 (p/v).
3.3.14 Solución de tiosulfato sódico al 8 por 100 (p/v).
3.3.15 Piedra pómez en granos, lavada con ácido clorhídrico y calcinada.
3.4 Procedimiento.
3.4.1 Mineralización.‒Pesar con precisión de 1 mg, aproximadamente, 1 de muestra e introducirla en el matraz de mineralización. Añadir 10 a 15 g de sulfato potásico, 0,3 a 0,4 g de catalizador óxido de cobre, 0,9 a 1,2 g de sulfato cúprico, 25 ml de ácido sulfúrico y algunos gránulos de piedra pómez. Homogeneizar. Calentar el matraz inicialmente con moderación, agitando de vez en cuando hasta carbonización de la masa y desaparición de espuma; calentar más intensamente hasta ebullición evitando el sobrecalentamiento y adherencia de partículas orgánicas.
Cuando la solución aparece transparente e incolora (verde claro en presencia de catalizador a base de cobre), mantener la ebullición una hora dejando enfriar a continuación.
3.4.2 Destilación.‒Añadir con precaución y agitando 250 a 350 ml de agua, comprobando que los sulfatos están disueltos totalmente. Dejar enfriar, añadir algunos gránulos de cinc y algunas gotas de indicador de fenolftaleína.
Introducir en el matraz colector del aparato de destilar 25 ml, exactamente medidos, de ácido sulfúrico 0,1 N o 0,5 N, según que el producto sea pobre o rico en materias nitrogenadas, y algunas gotas del indicador rojo de metilo.
Unir el matraz al refrigerante del aparato de destilar, sumergiendo la parte extrema de éste en el líquido del matraz colector, por lo menos, 1 cm. Introducir lentamente en el matraz, por medio de un embudo con llave, 120 ml de solución de hidróxido sódico al 30 por 100, o más cantidad si fuera necesario, debiéndose mantener la coloración roja hasta el fin de la destilación.
Calentar el matraz de tal manera que se destile 150 ml de líquido en treinta minutos. Después de este tiempo, comprobar la neutralidad del destilado por medio del papel de tornasol. Si la reacción es alcalina, continuar con la destilación hasta que el papel de tornasol indique neutralidad en la solución. Al final de la destilación, observar de vez en cuando la coloración de la solución en el colector. Si vira a amarillo, añadir inmediatamente un volumen exactamente medido de ácido sulfúrico 0,1 N o 0,5 N.
3.4.3 Valoración.‒Valorar en el matraz colector el exceso de ácido sulfúrico con la solución de hidróxido sódico 0,1 N o 0,25 N, según la normalidad del ácido sulfúrico utilizado hasta que la solución vire al amarillo claro.
3.5 Cálculos.

Siendo:
P = peso, en g, de la muestra.
V = solución de NaOH.
N = normalidad de la solución de ácido sulfúrico.
V' = volumen, ml, de NaOH consumidos en la valoración.
N' = normalidad de la solución de NaOH.
3.6 Observaciones.
3.6.1 Para productos pobres en materias nitrogenadas, introducir en el matraz colector 25 ml de ácido sulfúrico 0,1 N. Este volumen puede ser reducido, si es necesario, a 10 o 15 ml y completar a 25 ml con agua destilada.
3.6.2 Para los productos ricos en materias nitrogenadas, tales como las harinas de carne o de pescado, utilizar 25 ml de ácido sulfúrico 0,5 N.
3.7 Referencias.
1. Journal Officiel des Communautés Européennes. Número L 123/9.
4(a). GRASA BRUTA
(Sin hidrólisis previa)
4(a).1 Principio.
Extracción por éter dietílico de las materias grasas.
Aplicable a todos los piensos excepto los que figuran en 4(b).1.
4(a).2 Material y aparatos.
4(a).2.1 Extractor tipo Soxhlet o equivalente.
4(a).2.2 Aparato de calefacción con temperatura regulable, antideflagrante.
4(a).2.3 Estufa de desecación a vacío (menos de 100 Torr) (1 Torr = 1 mm Hg = 1/760 atm).
4(a).3 Reactivos.
4(a).3.1 Éter dietílico anhidro d = 0,720 p. e. = 34,5 °C, exento de peróxidos [4(a).6.1].
4(a).3.2 Sulfato sódico anhidro.
4(a).3.3 Tetracloruro de carbono.
4(a).4 Procedimiento.
4(a).4.1 Pesar con precisión de 1 mg, aproximadamente, 5 g de la muestra y mezclar con 2 o 3 g (o más, si es necesario) de sulfato sódico anhidro. Introducir la mezcla en un cartucho de extracción exento de materias grasas y recubrimiento de un tapón de algodón desengrasado.
Poner el cartucho en un extractor y extraer durante seis horas con el éter dietílico.
Regular el calor para obtener 15 extracciones o menos a la hora. Recoger el extracto etéreo en un matraz seco, conteniendo algunos fragmentos de piedra pómez y tarar. Reemplazar los fragmentos de piedra pómez por perlas de vidrio cuando la materia grasa debe ser objeto de análisis cualitativos posteriores.
Eliminar el éter por destilación e introducir el residuo durante una hora y media en la estufa de vacío a temperatura de 75 °C. Enfriar en desecador y pesar. Para comprobar que las pesadas de materia grasa son constantes, volver a introducir en la estufa de vacío durante treinta minutos.
4(a).4.2 Para los productos de contenido alto en materias grasas, difíciles de triturar o no apropiados para tomar una porción de una vez por no estar homogéneos, proceder como sigue: Pesar con precisión de 1 mg, aproximadamente, 20 g de muestra y mezclar con 10 g o más, de sulfato sódico anhidro. Proceder a la extracción con éter dietílico como se indica anteriormente. Redisolver el extracto en tetracloruro de carbono y llevarlo a un matraz aforado de 500 ml y homogeneizar. Tomar 50 ml de la solución y llevarla a un pequeño matraz seco, añadir unos trozos de piedra pómez y tarar. Eliminar el disolvente por destilación, secar y proseguir la operación como se indica anteriormente.
4(a).5 Cálculos.
Según 4(a).4.1.‒Expresar el resultado en porcentaje de la muestra.
Según 4(a).4.2.‒Expresar el resultado en porcentaje de muestra teniendo en cuenta los límites de parte alícuota utilizada en la primera extracción según la siguiente fórmula:
(10 a + b) • 5
Siendo:
a = extracto etéreo, en g, de la parte alícuota en la primera extracción.
b = extracto etéreo, en g, en la segunda extracción.
La diferencia entre los resultados de dos determinaciones paralelas efectuadas sobre la misma muestra no debe diferenciarse en más del 0,3 por 100 de materia grasa.
4(a).6 Observaciones.
4(a).6.1 Para el ensayo de los peróxidos, verter 10 ml de éter en una pequeña probeta tapada con tapón de vidrio, previamente enjuagada con éter, añadir 1 ml de solución al 10 por 100 de yoduro de potasio recién preparada, agitar y dejar reposar durante un minuto. No debe aparecer ningún color amarillo en ninguna de las capas. El éter dietílico podrá mantenerse exento de peróxidos añadiendo una lámina de cinc húmeda, que previamente deberá sumergirse completamente en una solución ácido diluida de sulfato de cobre durante un minuto y después lavar con agua. Utilizar por cada litro de éter una superficie de 80 cm2, aproximadamente, de lámina de cinc, cortada en bandas suficientemente largas para que lleguen, por lo menos, hasta la mitad del recipiente.
4(a).7 Referencias.
1. Journal Officiel des Communautés Européennes. Número L 279/17.
4(b). GRASA BRUTA
(Con hidrólisis previa)
4(b).1 Principio.
La muestra es hidrolizada en caliente por ácido clorhídrico, enfriada y filtrada. El residuo, lavado y secado, es sometido a la extracción por el éter dietílico según el procedimiento descrito en 4(a).
Aplicable a los productos cuyas materias grasas no puedan ser totalmente extraídas por el éter dietílico sin hidrólisis previa, como los productos de origen animal, gluten, pulpa seca de patatas, residuos de maltería y destilería, levadura seca, pan, productos de desecho de panadería, galletería y pastas alimenticias, productos lácteos y piensos enriquecidos.
4(b).2 Material y aparatos.
Como en 4 (a) 2.
4(b).3 Reactivos.
4(b).3.1 Como en 4(a).3.1.
4(b).3.2 Como en 4(a).3.2.
4(b).3.3 Ácido clorhídrico 3 N.
4(b).3.4 Material de filtración (tierra de diatomeas o similar).
4(b).4 Procedimiento
Pesar con precisión de 1 ml, aproximadamente, 2,5 g de la muestra e introducirlos en un vaso de 400 ml o en un Erlenmeyer de 300 ml. Añadir 100 ml de ácido clorhídrico 3 N y algunos trozos de piedra pómez. Tapar el vaso con un vidrio de reloj o acoplar al Erlenmeyer un refrigerante a reflujo. Llevar la mezcla a ebullición suave, con ayuda de una llama pequeña o una placa caliente y mantenerlo durante una hora, evitando que el producto se adhiera a las paredes del recipiente.
Enfriar y añadir una cantidad de material de filtración suficiente para evitar cualquier pérdida de materia grasa durante la filtración. Filtrar sobre un doble papel de filtro mojado, exento de materia grasa; lavar el residuo con agua fría hasta la desaparición de reacción ácida. Verificar que el filtrado no contiene materia grasa. La presencia de ésta en el filtrado indica que debe efectuarse una extracción de la muestra por el éter dietílico según se indica en 4(a).4.
Llevar el doble papel de filtro conteniendo el residuo sobre un vidrio de reloj y secar durante una hora y media en la estufa a temperatura de 95° - 98 °C.
Introducir el doble filtro y el residuo seco en un cartucho de extracción, extraer por el éter dietílico y proseguir el modo operatorio como en 4(a).4.
4(b).5 Cálculos.
Como en 4(a).5.
4(b).6 Referencias.
1. Journal Officiel des Communautés Européennes. Número 279/17.
5. CLORUROS
5.1 Principio
Los cloruros se solubilizan en agua, desecándose la solución si contiene materias orgánicas, posterior acidificación de la misma con ácido nítrico y precipitación de los cloruros con nitrato de plata. El exceso de nitrato se valora con una solución de sulfocianuro de amonio. Aplicable a todos los piensos.
5.2 Material y aparatos.
5.2.1 Agitador de 35 a 40 r.p.m.
5.3 Reactivos.
5.3.1 Solución de sulfocianuro de amonio 0,1 N.
5.3.2 Solución de nitrato de plata 0,1 N.
5.3.3 Solución saturada de sulfato amónico-férrico.
5.3.4 Ácido nítrico, d = 1,38.
5.3.5 Éter etílico.
5.3.6 Acetona.
5.3.7 Solución de Carrez I.‒Disolver en agua 24 g de acetato de cinc Zn (CH3COO)2 2H2O y 3 g de ácido acético glacial. Completar hasta 1.000 ml con agua.
5.3.8 Solución de Carrez II.‒Disolver en agua 10,6 g de ferrocianuro de potasio K4 [Fe (CN)6] • 3H2O. Completar a 100 ml con agua.
5.3.9 Carbón activo, exento de cloruros.
5.4 Procedimiento.
5.4.1 Preparación de la solución.
5.4.1.1 Muestras sin materia orgánica.‒Pesar, con precisión de 1 mg, de 0 a 10 g de la muestra de forma que no contenga más de 3 g de cloro en forma de cloruro e introducirla en un matraz aforado de 500 ml con 400 ml de agua a 20 °C aproximadamente. Agitar durante treinta minutos, enrasar, homogeneizar y filtrar.
5.4.1.2 Muestras con materia orgánica (menos los citados en 5.4.1.3).‒Pesar, con precisión de 1 mg, aproximadamente, 5 g de muestra e introducirla con 1 g de carbón activo en un matraz aforado de 500 ml. Añadir 400 ml de agua a 20 °C aproximadamente y 5 ml de solución de Carrez I. Agitar y añadir seguidamente 5 ml de la solución de Carrez II. Agitar durante treinta minutos, enrasar, homogeneizar y filtrar.
5.4.1.3 Torta y harina de lino, productos ricos en harina de lino y otros productos ricos en mucílagos o en sustancias coloidales (por ejemplo, almidón hidrolizado).‒Preparar la solución como se indica en 5.4.1.2, pero sin filtrar. Decantar (si es necesario centrifugar), separar 100 ml del líquido sobrenadante e introducirlos en un matraz de 200 ml. Mezclar con acetona y enrasar con este disolvente, homogeneizar y filtrar.
5.4.2 Valoración.‒Tomar de 25 a 100 ml de filtrado (con contenido en cloro inferior a 150 mg) obtenido en 5.4.1.1, 5.4.1.2 o 5.4.1.3 e introducirlo en un Erlenmeyer, diluir si es necesario, hasta 50 ml con agua. Añadir 5 ml de ácido nítrico, 20 ml de solución saturada de sulfato amónico férrico y dos gotas de la solución de sulfocianuro amónico, añadidas mediante una bureta llena hasta el trazo de cero. Añadir seguidamente mediante una bureta la solución de nitrato de plata mediante la solución de sulfocianuro amónico hasta que el viraje a rojo oscuro persista durante un minuto.
5.5 Cálculos
La cantidad de cloro (p), expresado en cloruro de sodio presente en el volumen del filtrado separado para la valoración, viene dada por la fórmula:
p = 5,845 (V1 ‒ V2) mg
Siendo:
V1 = volumen, en ml, de solución de nitrato de plata añadida.
V2 = volumen, en ml, de solución de sulfocianuro amónico 0,1 utilizados en la valoración.
Efectuar un ensayo en blanco sin la muestra a analizar y si consume solución de nitrato de plata 0,1 N restar este valor al volumen (V1 ‒ V2).
Expresar el resultado en porcentaje de la muestra.
5.6 Observaciones.
5.6.1 Para los productos ricos en materias grasas, desengrasar previamente mediante éter etílico según 4(a).
5.7 Referencias.
1. Journal Officiel des Communautés Européennes. Número L 155/23.
6. HUMEDAD
6.1 Principio.
La muestra se deseca en condiciones definidas, variando en función de la naturaleza del producto. La pérdida de masa es determinada por pesada. Es necesario proceder a una predesecación cuando se trata de sustancias sólidas, con un contenido elevado de humedad. No es aplicable a los productos derivados de la leche considerados como piensos simples, sustancias minerales, piensos compuestos constituidos esencialmente de sustancias minerales y semillas y frutos oleaginosos. Para los cereales y sus productos excepto productos de cereales hidrolizados y raicilla de cebada (6.3.2.2), se aplicará el método oficial para cereales y derivados.
6.2 Material y aparatos
6.2.1 Molino triturador, fácil de limpiar, que permita una trituración rápida y uniforme, sin provocar calentamientos sensibles, ni condensaciones, evitando al máximo el contacto con el aire.
6.2.2 Balanza analítica de precisión 0,5 mg.
6.2.3 Recipientes secos de metal inoxidable o de vidrio, provistos de una tapa que asegure un cierre estanco; superficie útil que permita obtener una distribución de la muestra del orden de 0,3 g por cm2.
6.2.4 Estufa isotérmica (± 1 °C) de calefacción eléctrica, que asegure una regulación rápida de temperatura y convenientemente ventilada.
6.2.5 Estufa de vacío, de calefacción eléctrica regulable, provista de una bomba de aceite o de un dispositivo de introducción de aire caliente deshidratado o de un deshidratante.
6.2.6 Desecador con placa de metal o porcelana, que contenga un deshidratante eficaz.
6.3 Procedimiento.
6.3.1 Preparación de la muestra.
6.3.1.1 Todas las muestras a excepción de las mencionadas en 6.3.1.2.
Separar previamente por lo menos 50 g de la muestra, triturándola o tratándola previamente de forma apropiada, si fuera necesario, para evitar toda variación del contenido en humedad.
6.3.1.2 Alimentos líquidos o pastosos, constituidos esencialmente por materias grasas.
Separar previamente y pesar, con aproximación de 10 mg, alrededor de 25 g de muestra. Añadir una cantidad apropiada de arena anhidra, pesada con aproximación de 10 mg y mezclar hasta obtener un producto homogéneo.
6.3.2 Desecación.
6.3.2.1 Todos los alimentos a excepción de los mencionados en 6.3.2.2.
Tarar, con aproximación de 0,5 mg, un recipiente provisto de tapa. Pesar, con aproximación de 1 mg, en el recipiente tarado unos 5 g de muestra y repartirla uniformemente. Colocar el recipiente, sin tapa, en una estufa previamente calentada a 103 ºC. Para evitar que la temperatura de la estufa no descienda demasiado, introducir el recipiente con rapidez. Dejar secar durante cuatro horas a partir del momento en que la estufa alcance de nuevo. La temperatura de 103 °C. Colocar la tapa sobre el recipiente, retirarlo de la estufa, dejar enfriar de 30 a 45 minutos en un desecador y pesar con aproximación de 1 mg.
En el caso de estar constituidos esencialmente por materias grasas, efectuar una desecación complementaria de treinta minutos en la estufa a 103 °C. La diferencia entre las dos pesadas no debe exceder de 0,1 por 100 de humedad.
6.3.2.2 Piensos compuestos que contengan más del 4 por 100 de sacarosa o de lactosa, productos de cereales hidrolizados, raicilla de cebada, garrofa, cabeza de remolacha, azúcares y solubles de pescado y piensos compuestos que contengan más del 25 por 100 de sales minerales con agua de cristalización.
Tarar, con una aproximación de 0,5 mg, un recipiente con tapa. Pesar, con una aproximación de 1 mg, en el recipiente tarado aproximadamente 5 g de muestra y repartirla uniformemente. Colocar el recipiente en la estufa de vacío previamente calentada a una temperatura de 80 a 85 °C, sin tapa. Para evitar que la temperatura de la estufa descienda demasiado, introducir el recipiente con rapidez. Llevar la presión a 100 Torrs, y dejar secar a esta presión durante cuatro horas, bajo una corriente de aire seco y caliente o con la ayuda de un deshidratante (300 g aproximadamente para 20 muestras). En este último caso, cortar la conexión con la bomba de vacío cuando la presión prescrita se alcance. Contar el tiempo de secado a partir del momento en que la estufa alcance de nuevo la temperatura de 80 a 85 °C. Llevar con precaución la estufa a la presión atmosférica.
Abrir la estufa, tapar inmediatamente el recipiente y sacarlo. Dejar enfriar durante cuarenta o cuarenta y cinco minutos en el desecador y pesar con una aproximación de 1 mg. Proceder a una desecación complementaria de 30 minutos en la estufa de vacío a la temperatura de 80 a 85 °C y pesar de nuevo. La diferencia entre las dos pesadas no debe exceder de 0,1 por 100 de humedad.
6.3.3 Predesecación.‒Los alimentos sólidos, cuyo contenido en humedad sea elevado y hagan la molienda difícil deben ser predesecados como se indica a continuación.
Pesar con una aproximación de 10 mg unos 50 g de muestra no molida (si es necesario puede hacerse una división previa en el caso de gránulos o aglomerados) en un recipiente adecuado.
Dejar secar en una estufa, a la temperatura de 60 a 70 °C, hasta que el contenido en humedad sea reducido a un valor comprendido entre 8 y 12 por 100.
Retirar de la estufa, dejar enfriar al aire en el laboratorio durante una hora y pesar con una aproximación de 10 mg. Triturar inmediatamente después como se indica en 6.3.1 y efectuar la desecación como en 6.3.2.1.
6.4 Cálculos.
El contenido en humedad, en tanto por ciento de muestra, se obtiene por las fórmulas siguientes:
6.4.1 Desecación sin predesecación.

Siendo:
M = masa inicial, en g, de la muestra.
m = masa, en g, de la muestra seca.
6.4.2 Desecación con predesecación.

Siendo:
E = masa inicial, en g, de la muestra.
M = masa en g, de la muestra predesecada.
M' = masa en g, de la muestra después de la molienda.
m = masa, en g, de la muestra seca.
La diferencia entre los resultados de dos determinaciones simultáneas efectuadas sobre una misma muestra no debe sobrepasar el 0,2 por 100 de humedad.
6.5 Referencias.
1. Journal Officiel des Communautés Européennes. Número L 279/8.
7. FIBRA BRUTA
7.1 Principio.
La muestra desengrasada se trata sucesivamente con soluciones de ácido sulfúrico e hidróxido potásico de concentraciones prefijadas. Separar el residuo filtrando sobre amianto, lavar, secar, pesar y calcinar a 900 °C. La pérdida de peso resultante de la calcinación corresponde a la fibra bruta.
7.2 Material y aparatos.
7.2.1 Vasos de 600 ml de capacidad graduados de 200 en 200 ml.
7.2.2 Placas filtrantes de porcelana de 80 mm de ∅, con un espesor de unos 4 mm, perforadas con 32 orificios de 4 mm de ∅ o similar.
7.2.3 Matraces aforados de 2 litros de capacidad con tapón de goma marcados con trazo de referencia al nivel de los 800 ml, provistos de un embudo de vidrio de 120 mm ∅.
7.2.4 Placas filtrantes, de 40 mm ∅ y 1 cm de espesor de 4 mm de borde oblicuo adaptado al cono del embudo (7.2.3), perforadas con 16 orificios de unos 4 mm de ∅ o similar y recubiertas de una rejilla metálica de 1 mm. Las placas y las rejillas metálicas deben ser inalterables a los ácidos y a los álcalis.
7.2.5 Crisoles de platino o cuarzo para la incineración.
7.2.6 Horno de mufla con termostato.
7.2.7 Desecador
7.2.8 Filtro de amianto.‒Poner 2 g de amianto en suspensión (7.3.2) en 100 ml de agua. Filtrar sobre placa filtrante recubierta de rejilla metálica (7.2.4) y colocada en el embudo del matraz aforado (7.2.3). Recoger el filtrado y filtrarlo de nuevo en el mismo filtro. Eliminar el filtrado.
7.2.9 Estufa.
7.3 Reactivos.
7.3.1 Ácido sulfúrico 0,26 N.
7.3.2 Amianto tratado.‒Añadir al amianto unas cinco veces su peso en ClH diluido (1 vol. de ClH d = 1,19 + 300 vol. de agua). Hervir la mezcla durante unos cuarenta y cinco minutos, dejar enfriar y filtrar. Lavar el residuo con agua hasta ausencia de acidez en el filtrado. Lavar a continuación con la acetona (7.3.6). Secar el amianto en la estufa y calcinar después durante dos horas a 900 °C. Dejar enfriar en desecador y conservar en un frasco cerrado.
7.3.3 Emulsión antiespuma.
7.3.4 Solución valorada de KOH 0,23 N.
7.3.5 Ácido clorhídrico 0,5 N.
7.3.6 Acetona pura.
7.3.7 Éter dietílico puro.
7.4 Procedimiento.
Pesar, con aproximación de 1 mg, 3 g de muestra y 2 g de amianto tratado en un vaso de 600 ml de capacidad. Añadir 200 ml de ácido sulfúrico 0,26 N y unas gotas de emulsión de antiespuma. Llevar rápidamente a ebullición y dejarla hervir durante treinta minutos exactos. Para mantener el volumen constante, cubrir el vaso con un dispositivo refrigerador, tal como un balón de fondo redondo de 500 ml sometido a una circulación de agua fría. Interrumpir la ebullición añadiendo 50 ml de agua fría y filtrar inmediatamente en un filtro de amianto.
Lavar el residuo con 100 ml de agua muy caliente, cinco veces, para obtener un volumen final de filtrado de 800 ml. Transferir cuantitativamente el residuo en el vaso provisto de un disco de porcelana para regular la ebullición. Añadir 200 ml de solución de hidróxido potásico 0,23 N. Llevar rápido a ebullición y dejar hervir durante treinta minutos exactos. Añadir 50 ml de agua fría y filtrar sobre un filtro de amianto nuevo preparado según 7.2 8. Lavar el residuo con agua muy caliente hasta la neutralidad de las aguas de lavado (con ayuda de papel de tornasol). Deshidratar lavando tres veces con acetona usando un volumen total de unos 100 ml.
Transferir cuantitativamente el residuo en un crisol, disgregarlo si es necesario, y secarlo a 130 °C hasta obtener un peso constante.
Dejar enfriar un desecador y pesar rápido. Introducir a continuación el crisol en el horno de mufla (7.2.6) y dejar calcinar durante treinta minutos a 900 °C. Dejar enfriar en desecador y pesar rápidamente.
Hacer un ensayo en blanco aplicando el mismo método operativo con amianto tratado (7.3.2) en ausencia de la muestra. La pérdida de peso resultante de la calcinación de los 6 g de amianto no debe ser superior a 6 mg.
7.5 Cálculos.
El contenido en fibra bruta en tanto por ciento de muestra viene dado por la fórmula:

Siendo:
a = pérdida de peso debida a la calcinación, en la muestra.
b = pérdida de peso debida a la calcinación, en el ensayo en blanco.
La diferencia entre dos determinaciones simultáneas efectuadas sobre la misma muestra no debe ser superior a:
0,3 en valor absoluto, para los contenidos en fibra bruta inferiores a 10 por 100.
3 por 100 en valor relativo para los contenidos en fibra bruta iguales o superiores al 10 por 100.
7.6 Observaciones.
7.6.1 Las muestras conteniendo más de un 10 por 100 de materia grasa deben desengrasarse con éter etílico antes del análisis. Para ello colocar la muestra (3 g ± 1 mg) sobre un filtro de amianto. Añadir unos 50 ml de éter dietílico y filtrar con vacío. Repetir la operación dos veces más. Transferir cuantitativamente la muestra desengrasada y el amianto a un vaso de 600 ml, continuando el análisis como se ha descrito anteriormente.
7.6.2 Las muestras con contenido en materias grasas protegidas por recubrimiento deben ser desengrasadas como se indica en 7.6.1 y ser sometidas, además, a un nuevo desengrasado después del lavado del residuo proveniente del ataque del ácido. Para esto lavar el residuo de este ataque tres veces con acetona (en total 100 ml), después tres veces con 50 ml de éter dietílico. Transferir a continuación cuantitativamente el residuo a un vaso de 600 ml y proseguir el análisis como se ha indicado anteriormente.
7.6.3 En el caso de muestras ricas en calcio (más de 2 por 100 de calcio) sustituir el tratamiento de ácido sulfúrico por el de ácido clorhídrico. Pesar 3 g ± 1 mg de muestra en un vaso y 2 g de amianto calcinado, introduciéndolos en un vaso de 600 ml. Añadir 100 ml de ClH 0,5 N y unas gotas de emulsión antiespuma. Dejar reposar durante cinco minutos. Proceder a continuación como se ha indicado anteriormente.
7.7 Referencias.
1. Journal Officiel des Communautés Européennes. Número L 83/24.
8. AZÚCARES
8.1 Principio.
Eliminación de todas las materias reductoras distintas de los azúcares, mediante defecación a partir de las soluciones de Carrez I, II, previa disolución de los azúcares en etanol diluido. Eliminación del etanol y valoración antes y después de la inversión según el método de Luff-Schoorl.
8.2 Material y aparatos.
8.2.1 Agitador mecánico.
8.2.2 Matraces aforados de 1.000, 300, 200, 100 y 50 ml.
8.3 Reactivos.
8.3.1 Etanol al 40 por 100 (v/v) d: 0,948 a 20 °C.
8.3.2 Solución de Carrez I.‒Disolver en agua 24 g de acetato de cinc y 3 g de ácido acético glacial y añadir agua destilada hasta 100 ml.
8.3.3 Solución Carrez II.‒Disolver en agua 10,6 g de ferro- cianuro potásico K4 (Fe CN6) 3H2O y añadir agua destilada hasta 100 ml.
8.3.4 Solución de rojo de metilo al 0,1 por 100 (v/v).
8.3.5 Ácido clorhídrico 4 N.
8.3.6 Ácido clorhídrico 0,1 N.
8.3.7 Solución de hidróxido sódico 0,1 N.
8.3.8 Solución de sulfato de cobre.‒Disolver 25 g de sulfato de cobre CuSO2 5H4O, exento de hierro, en agua y enrasar a 100 ml.
8.3.9 Solución de ácido cítrico.‒Disolver 50 g de ácido cítrico C6H8O7 H2O en 50 ml de agua.
8.3.10 Solución de carbonato sódico.‒Disolver 143,8 g de carbonato de sodio anhidro en 300 ml de agua caliente, dejar enfriar y completar a 300 ml.
8.3.11 Solución de tiosulfato sódico 0,1 N.
8.3.12 Solución de almidón.‒Añadir una mezcla de 5 g de almidón soluble en 30 ml de agua a un litro de agua hirviendo. Dejar hervir durante tres minutos. Dejar enfriar. Añadir 10 mg de ioduro mercúrico como agente conservado.
8.3.13 Ácido sulfúrico 6 N.
8.3.14 Solución de ioduro potásico al 30 por 100 (p/v).
8.3.15 Piedra pómez lavada con ácido clorhídrico y aclarada con agua.
8.3.16 Isopentanol.
8.3.17 Reactivo de Luff-Schoorl.‒Mezclar agitando lentamente 50 ml de la solución 8.3.10. Añadir en seguida 100 ml de la solución 8.3.8 y completar a un litro de agua. Dejar reposar doce horas y filtrar. Verificar la normalidad del reactivo obtenido (Cu 0,1 N; Na2Co32N). El pH de la solución debe ser aproximadamente 9,4.
8.4 Procedimiento.
8.4.1 Preparación de la muestra.‒Pesar con aproximación de 1 mg, 2,5 g de la muestra, e introducirlo en un matraz aforado de 250 ml. Añadir 200 ml de etanol al 40 por 100 (v/v) y mezclar durante una hora en el agitador. Añadir 5 ml de la solución Carrez I y agitar durante un minuto, adicionar y agitar durante el mismo tiempo con 5 ml de la solución Carrez II.
Enrasar a 250 ml con la solución de etanol (8.3.1), homogeneizar y filtrar. Tomar 200 ml del filtrado y evaporar aproximadamente hasta la mitad del volumen, a fin de eliminar la mayor parte del etanol. Transvasar en su totalidad el residuo de evaporación con ayuda de agua caliente a un matraz aforado de 200 ml y enfriar, a continuación enrasar con agua y filtrar si es necesario. Esta solución será utilizada para la determinación de azúcares reductores y después de la inversión para la determinación de azúcares totales.
8.4.2 Determinación de azúcares reductores.‒Tomar como máximo, 25 ml de la solución preparada según 8.4.1 y que contenga menos de 60 mg de azúcares reductores, expresado en glucosa. Si es necesario, completar el volumen hasta 25 ml con agua destilada y determinar la cantidad de azúcares reductores según Luff-Schoorl. El resultado será expresado en tantos por ciento de glucosa.
8.4.3 Determinación de azúcares totales previa inversión.‒Tomar 50 ml de la solución 8.4.1 y llevar a un matraz aforado de 100 ml. Añadir unas gotas de la solución rojo de metilo y adicionar lentamente agitando 15 ml de la solución de ácido clorhídrico 4 N hasta viraje a rojo. Añadir 15 ml de ácido clorhídrico 0,1 N y sumergirlo en un baño de agua caliente a ebullición durante treinta minutos. Refrigerar hasta 20 °C y añadir a continuación 15 ml de la solución de hidróxido sódico 0,1 N (8.3.7). Enrasar a 100 ml con agua y homogeneizar.
Tomar una cantidad que no exceda de 25 ml y contenga menos de 60 mg de azúcares reductores expresado en glucosa. Si es necesario, completar el volumen hasta 25 ml con agua destilada y determinar la cantidad de azúcares reductores según Luff-Schoorl. El resultado será expresado en tantos por ciento de glucosa. De expresarlo en sacarosa, se debe multiplicar por el factor 0,95.
8.4.4 Valoración de Luff-Schoorl.‒Tomar 25 ml del reactivo Luff-Schoorl (8.3.17) y llevarlo a un Erlenmeyer de 300 ml; añadir 25 ml exactamente medidos de la solución defecada de azúcares, adicionar un poco de piedra pómez y calentar agitando sobre la llama del mechero. Colocar inmediatamente el Erlenmeyer sobre una tela metálica, perforada por una abertura de 6 cm de diámetro y regulando la llama de manera que solamente el fondo del Erlenmeyer sea calentado. Adoptar en seguida un refrigerante de reflujo sobre el Erlenmeyer; a partir de este momento, hacer hervir la solución y mantener en ebullición durante diez minutos exactamente. Refrigerar inmediatamente al chorro de agua fría durante cinco minutos y proceder a su valoración.
Añadir 10 ml de la solución de ioduro potásico (8.3.14) inmediatamente después y con cuidado 25 ml de ácido sulfúrico 6 N (8.3.13). Valorar a continuación mediante la solución de tiosulfato de sodio 0,1 N (8.3.9) hasta la aparición de color amarillo, añadir en ese momento la solución de almidón y terminar de valorar.
Efectuar la misma valoración sobre una mezcla que contenga 25 ml exactamente medidos del reactivo de Luff-Schoorl, 25 ml de agua, 10 ml de la solución de ioduro de potasio (8.3.14) y 25 ml de la solución de ácido sulfúrico 6 N (8.3.13), sin llegar a ebullición.
8.5 Cálculos.
Establecer por medio de la tabla I la cantidad de glucosa en mg correspondiente a la diferencia entre las dos valoraciones, según los ml de tiosulfato de sodio 0,1 N gastados en cada una de las valoraciones.
Expresar los resultados en tanto por ciento de la muestra.
8.6 Observaciones.
8.6.1 En caso de alimentos muy ricos en melazas u otros alimentos poco homogéneos, pesar 20 g e introducirlos en un matraz aforado de un litro 500 ml de agua. Mezclar durante una hora en el agitador. Defecar mediante los reactivos de Carrez I y II (8.3.2 y 8.3.3), como se describe en 8.4.1, utilizando de todos los reactivos dosis cuatro veces superiores. Llenar a 1.000 ml con etanol al 40 por 100 (v/v) (8.3). Homogeneizar y filtrar; a continuación, eliminar el etanol según 8.4.1.
En ausencia de almidón exento de productos de hidrolizado, enrasar a 1.000 ml con agua destilada.
8.6.2 En el caso de melazas y alimentos simples, ricos en azúcares y prácticamente exentos de almidón, pesar 5 g e introducirlos en un matraz aforado de 250 ml, añadir 200 ml de agua destilada y mezclar durante una hora o más en el agitador. Defecar inmediatamente por medio de los reactivos de Carrez i y II (8.3.2 y 8.3.3), según 8.4.1
Llevar a 250 mí con agua, homogeneizar y filtrar, para determinar los azúcares totales, proseguir como 8.4.3.
8.6.3 Es recomendable añadir, aproximadamente, 1 ml de isopentanol (sin tener en cuenta el volumen) antes de la ebullición, con el reactivo Luff-Schoorl para evitar la formación de espuma.
8.6.4 La diferencia entre la cantidad de azúcares totales después de la inversión, expresada en glucosa, y la cantidad de azúcares reductores, expresada igualmente en glucosa, multiplicada por 0,95, da la cantidad en tanto por ciento de sacarosa.
8.6.5 Para calcular la cantidad de azúcares reductores, excluyendo la lactosa, se puede determinar de las siguientes formas:
8.6.5.1 Para un cálculo aproximado, multiplicar por 0,675 la cantidad de lactosa obtenida, por determinación separada, y restar el resultado obtenido de la cantidad en azúcares reductores.
8.6.5.2 Para el cálculo preciso de azúcares reductores, excluyendo la lactosa, es necesario partir de la misma muestra (8.4.1) para las dos determinaciones finales. Uno de los análisis es efectuado a partir de la solución obtenida en 8.4.1 y el otro sobre una parte de la solución obtenida para la valoración de la lactosa según el método para la determinación de lactosa.
En los casos 8.6.5.1 y 8.6.5.2, la cantidad de azúcares presentes se determinan según el método de Luff-Schoorl, expresado en mg de glucosa.
La diferencia entre los dos valores se expresa en tanto por ciento de la muestra.
8.7 Bibliografía.
1. Journal Officiel des Communautés Européennes. Número L 155/32.
TABLA I
Para 25 ml de reactivo Luff-Schoorl
|
Na2S2O3 0,1 N |
Glucosa, fructosa Azúcares invertidos C6H12O6 |
Lactosa C12H22O11 |
Maltosa C12H22O11 |
|||
|---|---|---|---|---|---|---|
| ml | mg | Diferencia | mg | Diferencia | mg | Diferencia |
| 1 | 2,4 | 2,4 | 3.6 | 3,7 | 3,9 | 3,9 |
| 2 | 4,8 | 2,4 | 7,3 | 3,7 | 7,8 | 3,9 |
| 3 | 7,2 | 2,5 | 11,0 | 3,7 | 11,7 | 3,9 |
| 4 | 9,7 | 2,5 | 14.7 | 3,7 | 15,6 | 4,0 |
| 5 | 12,2 | 2,5 | 18 4 | 3,7 | 19,6 | 3,9 |
| 6 | 14 7 | 2,5 | 22 I | 3,7 | 23,5 | 4,0 |
| 7 | 17,2 | 2 6 | 25,8 | 3,7 | 27,5 | 4,0 |
| 8 | 19,8 | 2,6 | 29,5 | 3,7 | 31,5 | 4,0 |
| 9 | 22,4 | 2,6 | 33,2 | 3,8 | 35,5 | 4,0 |
| 10 | 25,0 | 2,6 | 37,0 | 3,8 | 39,5 | 4,0 |
| 11 | 27,6 | 2,7 | 40,8 | 3,8 | 43,5 | 4,0 |
| 12 | 30,3 | 2,7 | 44,6 | 3,8 | 47,5 | 4,1 |
| 13 | 33,0 | 2,7 | 48,4 | 3,0 | 51,6 | 4,1 |
| 14 | 35,7 | 2,8 | 52,2 | 3,8 | 55,7 | 4,1 |
| 15 | 38,5 | 2,8 | 56,0 | 3,9 | 59,8 | 4,1 |
| 16 | 41,3 | 2,9 | 59,9 | 3,9 | 63,9 | 4,1 |
| 17 | 44,2 | 2,9 | 63,8 | 3,9 | 68,0 | 4,2 |
| 18 | 47,1 | 2,9 | 67,7 | 4,0 | 72,2 | 4,3 |
| 19 | 50,0 | 3,0 | 71,7 | 4,0 | 76,5 | 4,4 |
| 20 | 53,0 | 3,0 | 75,7 | 4,1 | 80,9 | 4,5 |
| 21 | 56,0 | 3,1 | 79,3 | 4,1 | 85,4 | 4,6 |
| 22 | 59,1 | 3,1 | 83,9 | 4,1 | 90,0 | 4,6 |
| 23 | 62,2 | 88,0 | 94,6 | |||
9. ACIDEZ DE LA GRASA
9.1 Principio.
Las materias grasas son extraídas por éter dietílico y subsiguiente neutralización con hidróxido sódico.
9.2 Material y aparatos.
9.2.1 Extractor tipo Soxhlet o equivalente.
9.2.2 Aparato de calefacción a temperatura regulable, antideflagrante.
9.2.3 Estufa de desecación a vacío (menos de 100 Torr) (1 Torr = 1 mmHg = 1/760 atm).
9.3 Reactivos.
9.3.1 Éter dietílico anhidro d = 0,720, p. e., 34,5 °C, exento de peróxidos 14 (a).6.11.
9.3.2 Sulfato sódico, anhidro.
9.3.3 Ácido clorhídrico 3 N.
9.3.4 Material de filtración; por ejemplo, tierra de diatomeas.
9.3.5 Tetracloruro de carbono.
9.3.6 Alcohol etílico.
9.3.7 Solución de hidróxido sódico 0,1 N.
9.4 Procedimiento.
9.4.1 Pesar, con aproximación de 1 mg, 5 g de la muestra y mezclar con 2 o 3 g (o más, si es necesario) de sulfato sódico anhidro. Introducir la mezcla en un cartucho de extracción exento de materias grasas y recubierto de un tapón de algodón desengrasado.
Poner el cartucho en un extractor y extraer durante seis horas con éter dietílico.
Regular el calor para obtener 15 extracciones o menos a la hora. Recoger el extracto etéreo en un matraz seco, conteniendo algunos fragmentos de piedra pómez y tarar. Reemplazar los fragmentos de piedra pómez por perlas de vidrio cuando la materia grasa debe ser objeto de análisis cualitativos posteriores.
Eliminar el éter por destilación y secar seguidamente el residuo a evaporación durante una hora y media en la estufa de desecación a vacío a temperatura de 75 °C. Enfriar en desecador y pesar. Efectuar una segunda desecación durante 30 minutos para asegurar que las pesadas de materia grasa son constantes (la diferencia de pesadas debe ser inferior a 1 mg).
9.4.2 Para los productos de contenido alto en materias grasas, difíciles de triturar o no apropiados para tomar una porción de una vez por no estar homogéneos, proceder como sigue: Pesar, con la aproximación de 1 mg, 20 g de muestra y mezclar con 10 g o más de sulfato sódico anhidro. Proceder a la extracción con éter dietílico como se indica anteriormente. Redisolver el extracto en tetracloruro de carbono y llevarlo a un matraz aforado de 500 ml y homogeneizar. Tomar 50 ml de la solución y llevarla a un pequeño matraz seco, añadir unos trozos de piedra pómez y tarar. Eliminar el disolvente por destilación, secar y proseguir la operación como se indica anteriormente. Eliminar el disolvente del residuo de extracción que se encuentra en el cartucho y moler el residuo con finura de 1 mm. Colocar de nuevo el producto en el cartucho de extracción (no añadir sulfato sódico), extraer con éter dietílico y proseguir la operación como se indica anteriormente.
Disolver el extracto etéreo con 15 ml de una mezcla a volúmenes iguales de éter y alcohol dietílico de 96º y valorar con hidróxido sódico N/10 de factor conocido empleando fenolftaleína como indicador hasta color rosa persistente. Se hace un testigo en blanco para restarle la acidez de la mezcla alcohol-etérea.
9.5 Cálculo.
Expresar el resultado en tanto por ciento de acidez en ácido oleico:

Siendo:
V = volumen, en ml, de NaOH N/10 gastados, restándole los gastados en la prueba testigo.
P = peso de grasa obtenido en la extracción,
9.6 Observaciones.
9.6.1 Para el ensayo de los peróxidos verter 10 ml de éter en una pequeña probeta tapada con tapón de vidrio, previamente enjuagada con éter, añadir 1 ml de solución al 10 por 100 de yoduro de potasio, recién preparada. Agitar y dejar reposar durante un minuto No debe aparecer ningún color amarillo en ninguna de las capas. El éter dietílico podrá mantenerse exento de peróxidos añadiendo una lámina de cinc húmeda, que deberá sumergirse completamente en una solución ácida diluida de sulfato de cobre durante un minuto y después lavar con agua. Utilizar por litro una superficie de 80 cm2 aproximadamente de lámina de cinc, cortada en bandas suficientemente largas para que lleguen por lo menos hasta la mitad del recipiente.
9.6.2 En harinas de pescado empléese como indicador azul alcalino 6-B disuelto al 0,5 por 100 en etanol.
10. CALCIO
10.1 Principio.
Incineración de la muestra y precipitación del calcio mediante ácido clorhídrico en forma de oxalato cálcico y valoración del ácido oxálico formado con una solución de permanganato potásico previa disolución del precipitado con ácido sulfúrico. Aplicable a los alimentos de animales.
10.2 Material y aparatos.
10.2.1 Matraz aforado de 250 ml de capacidad.
10.2.2 Erlenmeyer de 250 ml de capacidad.
10.2.3 Crisol de platino, cuarzo o porcelana.
10.2.4 Crisoles filtrantes de vidrio, porosidad G4.
10.2.5 Horno eléctrico con circulación de aire y regulador automático, para regular a 550 °C.
10.2.6 Baño de agua.
10.3 Reactivos.
10.3.1 Ácido clorhídrico d: 1,14.
10.3.2 Ácido nítrico d: 1.40.
10.3.3 Ácido sulfúrico d: 1,13.
10.3.4 Amoníaco d: 0,98.
10.3.5 Solución saturada de oxalato amónico.
10.3.6 Solución al 30 por 100 (p/v) de ácido cítrico.
10.3.7 Solución al 5 por 100 (p/v) de cloruro de amonio.
10.3.8 Solución al 0,04 por 100 (p/v) de verde de bromocresol.
10.3.9 Solución de permanganato potásico: 0,1 N.
10.4 Procedimiento.
Pesar, con aproximación de 1 mg, 5 g de la muestra a analizar (o más si es necesario), calcinarla a 550 °C y transvasar las cenizas a un Erlenmeyer de 250 ml. Añadir 40 ml de ácido clorhídrico (10.3.1), 60 ml de agua y algunas gotas de ácido nítrico (10 3.2). Llevar a ebullición y mantenerlo así durante treinta minutos Enfriar, transvasar la solución a un matraz aforado de 250 ml, enjuagar el Erlenmeyer y completar el volumen con agua, homogeneizar y filtrar.
Tomar con una pipeta según la cantidad presumible de calcio, una alícuota que contenga de 10 a 40 mg de calcio e introducirla en un Erlenmeyer de 250 ml. Añadir 1 ml de la solución de ácido cítrico (10.3.6) y 5 ml de solución de cloruro de amonio (10.3.7). Completar el volumen a 100 ml aproximadamente con agua. Llevar a ebullición, añadiendo de 8 a 10 gotas de solución de verde de bromocresol (10.3.8) y 30 ml de solución caliente de oxalato de amonio (10.3.5). Si aparece un precipitado, disolver éste mediante la adición de algunas gotas de ácido clorhídrico r10.3 1).
Neutralizar en seguida muy lentamente con amoníaco (10.3.4), agitando constantemente, hasta obtener un pH 4,4-4,6 (viraje del indicador). Colocar el Erlenmeyer en un baño de agua hirviendo durante treinta minutos, dejando reposar el precipitado formado Retirarlo del baño, dejarlo reposar durante una hora y filtrar en un crisol filtrante Gμ. Lavar el Erlenmeyer y el crisol con agua hasta la total eliminación del exceso de oxalato de amonio (la ausencia de cloruros en el agua de lavado indica que el lavado es suficiente).
Disolver el precipitado sobre el filtro con 50 ml de ácido sulfúrico caliente (10.3.3), enjuagar el crisol con agua caliente hasta llevar el filtrado a 10 ml aproximadamente. Calentar a 70-80 °C y valorar mediante la solución de permanganato potásico (10.3.9) hasta la obtención de una coloración rosa persistente durante un minuto.
10.5 Cálculo.
Un ml de permanganato potásico 0,1 N corresponde a 2,004 mg de calcio.
Expresar el resultado obtenido en tanto por ciento de la muestra.
10.6 Observaciones.
10.6.1 Para pequeñas cantidades de calcio, proceder como en 10.5. Filtrar el precipitado de oxalato de calcio sobre un papel de filtro sin cenizas y calcinarlo en un crisol a 550 °C. Recuperar el residuo con algunas gotas de ácido sulfúrico (10.3.3), evaporar a sequedad, calcinar de nuevo a 550 °C y pesar.
Si P representa el peso de sulfato cálcico obtenido, la cantidad en calcio de la alícuota tomada será igual a P x 0,2944.
10.6.2 Si la muestra está constituida exclusivamente de materias minerales, proceder a la disolución por ácido clorhídrico sin incineración previa. Para los productos tales que los fosfatos alumínico-cálcicos difíciles de disolver en los ácidos, proceder a una fusión alcalina antes de la disolución. Mezclar íntimamente en un crisol de platino la parte tomada con cinco veces su peso de una mezcla compuesta en partes iguales de carbonato de potasio y de carbonato de sodio. Calentar con precaución hasta la fusión completa de la mezcla, refrigerar y disolver con ácido clorhídrico.
10.6.3 Si la cantidad en magnesio de la muestra es elevada, proceder a una segunda precipitación con oxalato de calcio.
10.7 Referencias.
I Journal Officiel des Communautés Européennes. Número L 155/17.
11. GRASA
(En productos lácteos reengrasados)
11.1 Principio.
Determinación de la grasa por el método Gerber.
11.2 Material y aparatos.
11.2.1 Butirómetros contrastados, con graduaciones de 0-6 % de grasa y con divisiones del 0,1 %. Se pueden utilizar butirómetros con graduaciones de 0 a 5 % y divisiones del 0,1 % cuando muestra tenga un contenido en grasa inferior al 5%.
11.2.2 Tapones troncocónicos de goma u otro tipo, apropiados.
11.2.3 Pipetas contrastadas de 1 y 10 ml.
11.2.4 Vidrios de reloj.
11.2.5 Baño de agua regulable a 65° ± 1°C.
11.2.6 Centrífuga capaz de alcanzar 1.200 r.p.m.
11.2.7 Embudo desprovisto de cuello y vástago para la introducción de la muestra en el butirómetro.
11.3 Reactivos.
11.3.1 Ácido sulfúrico de densidad d = 1,82.
11.3.2 Alcohol isoamílico de densidad d = 0,815 e intervalo de destilación de 128° a 132 °C.
11.4 Procedimiento.
11.4.1 Pesar, con precisión de 0,1 mg, 1.100 g de producto e introducirlo en el butirómetro a través del embudo. Añadir, a través del embudo, 10 ml de agua tibia (40 °C), arrastrando lo que haya podido adherirse al mismo. Tapar y agitar fuertemente para disolver la leche.
Seguidamente añadir 10 ml de ácido sulfúrico haciéndolo resbalar suavemente por las paredes del butirómetro. Añadir 1 ml de alcohol isoamílico. Cerrar el butirómetro con el tapón de goma, agitar suavemente y centrifugar a 1.200 r.p.m. durante tres a cinco minutos. Sacar el butirómetro de la centrífuga e introducir en el baño de agua a 65 °C. Dejar transcurrir algunos minutos y efectuar la lectura.
11.5 Expresión de los resultados.
Leer en el butirómetro, en la escala dividida, la altura que ha alcanzado la columna de grasa, habiéndose ajustado a cero el líquido que no contiene grasa. Los grados de la escala indican las decenas, y las décimas, las unidades por ciento. Se puede apreciar perfectamente un 0,5 por 100.
11.6 Observaciones.
11.6.1 En el caso de sueros desprovistos de caseína, pueden producir carbonizaciones que dificultan la lectura (debido a que el ácido sulfúrico, de densidad d = 1,82, resulta demasiado concentrado). En este caso se puede diluir al 10 por 100 (v/v) con agua destilada.
11.6.2 En los productos con grasa muy micronizada conviene repetir por tres veces las centrifugaciones, el calentamiento a 65 °C y las lecturas, hasta obtener resultados constantes.
11.7 Referencias.
1. Instituto de Racionalización y Normalización del Trabajo. Una Norma Española 64.029.
12. CENIZAS BRUTAS
12.1 Principio.
Incineración de la muestra a 550 °C y pesada del residuo hasta peso constante.
12.2 Material y aparatos.
12.2.1 Placa calefactora.
12.2.2 Horno eléctrico con regulación de temperatura.
12.2.3 Crisoles de platino o cuarzo, rectangulares (60 x 40 x 25 mm) o redondos.
12.2.4 Nitrato de amonio.‒Solución al 20 por 100.
12.3 Procedimiento.
Pesar alrededor de 5 g de muestra, con una aproximación de 1 mg (para los productos que tengan tendencia a «esponjarse», pesar 2,5 g) en un crisol previamente calcinado y tarado. Colocar el crisol sobre la placa calefactora hasta carbonización de la muestra. Introducir el crisol en el horno regulado a 550 ± 5 °C. Mantener a esta temperatura hasta la obtención de cenizas blancas, gris claro o rojizas, aparentemente desprovistas de partículas carbonosas. Colocar el crisol en un desecador, dejar enfriar y pesar inmediatamente.
12.4 Cálculo.
El porcentaje de cenizas sobre materia natural se obtiene por la fórmula siguiente:

Siendo:
P = peso, en g, de la cápsula con la muestra.
P1 = peso, en g, de la cápsula con las cenizas.
P2 = peso, en g, de la cápsula vacía.
12.5 Observaciones.
12.5.1 Las materias difíciles de incinerar deben someterse a una primera incineración de tres horas, se enfrían y se les adiciona algunas gotas de una solución al 20 por 100 de nitrato de amonio. Continuando la incineración después de la desecación en estufa.
Repetir eventualmente la operación hasta incineración completa.
12.5.2 Para las materias resistentes al tratamiento anterior, operar como sigue: Después de una incineración de tres horas, arrastrar las cenizas con agua caliente y filtrar sobre un pequeño filtro de cenizas conocidas. Incinerar el filtro y su contenido en el crisol inicial.
Llevar el filtrado al crisol frío, evaporar a sequedad, incinerar y pesar.
12.5.3 En el caso de aceites y grasas, pesar 25 g en un crisol de capacidad apropiada. Carbonizar inflamando la muestra por medio de una mecha de papel de filtro sin cenizas. Después de la combustión, humedecer con la mínima cantidad de agua posible. Desecar y continuar como se indica en 12.5.
12.6 Referencias.
1. Journal Officiel des Communautés Européennes. Número L 155/20.
1. BIÓXIDO DE CARBONO LIBRE (CO2)
1.1 Principio.
El presente método está basado en la reacción del CO2 libre del agua con el hidróxido de sodio para formar bicarbonato de sodio.
Además de este método de valoración volumétrica existe uno monográfico que por exigir gran precisión en la determinación «in situ» del pH y de la alcalinidad resulta menos aconsejable.
Pueden producirse errores por la presencia de amoníaco, aminas, fosfatos, boratos, silicatos, sulfuros y nitritos, así como por la existencia de ácidos minerales o sales de ácido fuerte y base débil. También alteran cuantitativamente el resultado el aluminio, hierro, cromo y cobre.
1.2 Material y aparatos.
1.2.1 Frascos pyrex de 1.000 ml.
1.2.2 Probeta graduada de 100 ml.
1.2.3 Tubo de goma.
1.2.4 Bureta normal graduada en 0,1 ml.
1.2.5 Varilla de vidrio.
1.3 Reactivos.
1.3.1 Solución valorada de hidróxido sódico 0,02 N.‒Diluir 20 ml de NaOH 1 N a un litro de agua destilada que previamente se ha hervido no menos de quince minutos para expulsar el bióxido de carbono y enfriarlo a la temperatura ambiente. Preparar diariamente y proteger del bióxido de carbono atmosférico en un frasco pyrex. Titular la solución con el reactivo 1.3.3.
1.3.2 Solución indicadora de fenolftaleína. Disolver 0,5 g de fenolftaleína en 50 ml de alcohol etílico de 96° y agregar 50 ml de agua destilada que previamente se ha hervido no menos de quince minutos para expulsar el bióxido de carbono y enfriarlo a la temperatura ambiente. Agregar a continuación el reactivo 1.3.1 a gotas hasta que aparezca una muy ligera coloración rosa.
1.3.3 Solución patrón ácida 0,01 N de ácido clorhídrico o sulfúrico.
1.4 Procedimiento.
Sifonar la muestra mediante tubo de goma, llenando desde el fondo una probeta de 100 ml hasta que derrame. Extraer el tubo de goma y verter el exceso de agua mediante sacudidas.
Agregar cinco o diez gotas del reactivo 1.3.2. Si la muestra vira a rojo, no hay presente bióxido de carbono libre, si la muestra se mantiene incolora, titular rápidamente con el reactivo 1.3.1 agitando suavemente con una varilla de vidrio hasta que el color rosa característico observado a través de todo el espesor de la muestra persista, por lo menos, treinta segundos.
1.5 Cálculos.
Calcular el bióxido de carbono libre, expresado en miligramos por litro, mediante la fórmula.

Siendo:
A = ml gastados del reactivo 1.3.1.
F = factor de corrección del reactivo 1.3.1 hallado mediante su titulación con el reactivo 1.3.3.
B = ml de muestra.
1.6 Referencias.
1. Métodos de análisis recomendados. Centro de Estudios Hidrográficos. Madrid, 1968.
2. Standard Methods for the Examination of Water and Wastewater. American Public Health Association, Inc. New York, 1960.
2. DEMANDA BIOQUÍMICA DE OXÍGENO (D. B. O.)
2.1 Principio.
La demanda bioquímica de oxígeno o D. B. O., es la cantidad de oxígeno eventualmente consumida por los gérmenes aerobios para asegurar la descomposición, en condiciones normalizadas de incubación, de las materias orgánicas contenidas en el agua analizada.
La incubación se efectúa en oscuridad, durante cinco días, y a una temperatura de 20 °C.
2.2 Material y aparatos.
2.2.1 Frascos de incubación, de 250-300 ml de capacidad; aforados y con tapón esmerilado macizo y con bisel.
2.2.2 Estufa de cultivos con control termostático a 20 °C ± 1 ºC.
2.2.3 Matraces Erlenmeyer de 500 ml.
2.2.4 Pipetas de 1 y 5 ml.
2.2.5 Buretas.
2.3 Reactivos.
2.3.1 Agua destilada.
2.3.2 Solución amortiguadora de fosfato.‒Disolver 8,5 g de KH2PO4, 25,75 g de K2HPO4, 33,4 g de Na2HPO4, 7H2O y 1,7 g de NH4Cl en unos 500 ml de agua destilada y diluir a un litro. El pH de esta solución amortiguadora debe ser de 7,2, sin juste alguno.
2.3.3 Solución de sulfato de magnesio.‒Disolver 22,5 g de MgSO4 • 7H2O en agua destilada y diluir a un litro.
2.3.4 Solución de cloruro de calcio.‒Disolver 27,5 g de CaCl2 anhidro en agua destilada y diluir a un litro.
2.3.5 Solución de cloruro férrico.‒Disolver 0,25 g de FeCl3 6H2O en agua destilada y diluir a un litro.
2.3.6 Soluciones de ácidos y álcalis 1 N.
2.3.7 Solución de sulfito de sodio 0,025 N.‒Disolver 1,575 g de NaSO3 anhidro en 1.000 ml de agua destilada. Esta solución no es estable y se debe preparar el día que se vaya a emplear.
2.3.8 Solución de sulfato manganoso.‒Disolver 400 g de MnSO4 • 2H2O en agua destilada, filtrar y diluir a un litro. Esta solución no debe liberar más que trazas de yodo cuando se añade a una solución acidificada de ioduro potásico.
2.3.9 Reactivo de álcali-ioduro nitruro.‒Disolver 500 g de NaOH y 135 g de Nal en agua destilada Disolver 10 g de NaN3 en 40 ml de agua destilada. Mezclar ambas soluciones y diluir a un litro. Este reactivo no debe producir coloración con el almidón cuando se diluye y acidifica.
2.3.10 Ácido clorhídrico concentrado de densidad 22° Beaumé = 1,18.
2.3.11 Solución de almidón.‒Triturar 5 g de almidón soluble y 5 mg de I2Hg con un poco de agua y añadir lentamente la emulsión a un litro de agua hirviendo, manteniendo la ebullición unos minutos hasta que la solución quede clara. Enfriar y pasar a un frasco topacio con tapón esmerilado.
2.3.12 Solución Stock de tiosulfato sódico 0,10 N.‒Disolver 24,82 g de Na2S2O3 • 5H2O en agua destilada recién hervida y enfriada y diluir a un litro. Conservar la solución añadiendo 1 g de NaOH.
2.3.13 Solución valorada de dicromato potásico 0,025 N.‒Pesar 1,226 g de K3Cr2O7, previamente secados durante dos horas a 103 °C y diluir a un litro en matraz aforado.
2.3.14 Solución valorada de tiosulfato sódico 0,025 N.‒Diluir 250 ml del reactivo 2.3.12 a un litro de agua destilada recientemente hervida y enfriada. Titular esta solución mediante el reactivo 2.3.13.
2.4 Procedimiento.
2.4.1 Preparación del agua de dilución.
Tomar dos litros de agua destilada.
Oxigenarla mediante aire comprimido hasta que la tensión del oxígeno disuelto esté próxima a su saturación.
Añadir a dicha agua ü ml de cada uno de los reactivos 2.3.2, 2.3.3, 2.3.4 y 2,3 5.
Conservar este agua a 20 °C, tapado el frasco con tapón de algodón.
2.4.2 Acondicionamiento del agua problema.
Neutralizar el agua problema hasta pH próximo a 7 mediante el reactivo 2.3.6.
Eliminar el cloro libre, si lo hubiese, mediante la adición del reactivo 2.3.7.
Eliminar el exceso de oxígeno agitando fuertemente la muestra o bien mediante burbujeo de aire comprimido, siempre a 20 °C.
2.4.3 Dilución del agua problema.
Sifonar el agua de dilución a una probeta graduada de 1.000-2.000 ml de capacidad, llenándola hasta la mitad sin arrastrar aire. Agregar el agua de muestra cuidadosamente mezclada en la cantidad necesaria para obtener la dilución que se desee y terminar de diluir hasta el nivel apropiado con agua de dilución. Mezclar bien con un agitador, evitando el arrastre de aire.
Se aconseja las siguientes diluciones:
Efluentes depurados: de 5 a 25 por 100.
Aguas fluviales: de 25 a 100 por 100.
Sifonar la dilución mezclada a dos frascos de incubación, llenándolos completamente y cerrándolos herméticamente.
Determinar el oxígeno inicial en uno de los dos frascos de forma inmediata y conservar el otro frasco en incubación durante cinco días en oscuridad y a una temperatura de 20 °C, para después determinar en él el oxígeno disuelto no consumido.
2.4.4 Determinación del oxígeno disuelto:
Agregar al frasco de incubación 1 ml del reactivo 2.3.8 y después l ml del reactivo 2.3.9, haciendo ambas adiciones en el fondo del frasco. Tapar de nuevo el frasco procurando excluir las burbujas, con lo que se derramará una pequeña cantidad de líquido.
Mezclar por inversión varias veces y esperar que se sedimente el precipitado.
Destapar el frasco y añadir, mediante pipeta, haciendo llegar el ácido hasta el fondo, 5 ml de 2.3.10. Cerrar de nuevo el frasco y agitar hasta que se disuelva el precipitado.
Pasar todo el líquido a un matraz Erlenmeyer de 500 ml, lavando frasco y tapón con agua destilada.
Añadir gota a gota, con rapidez y agitando el matraz, el reactivo 2 3.14 hasta que el líquido tome coloración paja pálido.
Agregar 1 o 2 ml del reactivo 2.3.11, con lo que tomará el líquido color azul, y continuar valorando con 2.3.14, gota a gota, hasta que el líquido se torne incoloro o gris blanquecino.
2.5 Cálculos.
Calcular el oxígeno disuelto, expresado en p. p. m. mediante la fórmula:

Siendo:
V = ml de tiosulfato gastados.
f = factor de corrección de la normalidad del tiosulfato hallado mediante la titulación con reactivo 2.3.13.
A = volumen, en ml, del frasco utilizado.
Calcular la D B. O., expresada en p. p. m., mediante la fórmula:

Siendo:
D1 = p. p. m. de O2 disuelto en el frasco no sometido a incubación.
D2 = p. p. m. de O2 disuelto en el frasco sometido a incubación.
C = tanto por ciento del agua problema en el agua problema diluida.
2.6 Observaciones.
Este método no es aplicable a aguas negras o desechos industriales concentrados que exigen diluciones muy elevadas y obligan a sembrar el agua de dilución con un inóculo.
En estos casos se precisan comprobaciones de la D. B. O. del inóculo y los métodos y cálculos requieren técnicas laboriosas sólo propias de los laboratorios especializados.
2.7 Referencias.
1. Standard Methods for the Examination of Water and Sewage. American Public Health Association. New York, 1960.
2. Rodier, J.: L’Analyse chimique et phisico-chimique de l’eau. Dunod. París.
3. Gutiérrez Calderón, E., et alt.: Estudio del B. O. D. y plan de trabajo para su determinación. Anales I. F. I. E. Madrid, 1959.
4. Livre de l’eau. Cebedoc. Lieja, 1966.
3. CONDUCTIVIDAD ELÉCTRICA
3.1 Principio.
Se denomina conductividad específica de un agua a la aptitud de ésta para transmitir la corriente eléctrica.
La conductividad depende de la actividad y tipo de iones disueltos y de la temperatura a la que se realiza la medida.
Para medir la conductividad se hace uso de un puente de Wheatstone y una célula de conductividad apropiada, comparando a la misma temperatura la resistencia eléctrica de la muestra y de una solución valorada de cloruro potásico y refiriendo el resultado a 25 °C.
3.2 Material y aparatos.
3.2.1 Puente de Wheatstone apto para este fin.
3.2.2 Celda de conductividad específica, bien del tipo pipeta, bien del tipo inmersión, con electrodos de platino platinados La constante de la celda debe ser aproximadamente de 10-1 para aguas de conductividad baja, de 1 para aguas de conductividad media y de 10 para aguas de conductividad alta.
3.3 Reactivos.
Solución patrón de ClK.‒Preparar una solución de ClK cuya conductividad difiera por un factor de menos de 5 de la conductividad de la muestra. Generalmente, una solución 0,01 M de ClK (0,7456 g/l) es aceptable. Esta solución tiene una conductividad de 1.413 μmho/cm. De la siguiente tabla pueden elegirse otras soluciones patrones que se consideren necesarias.
Conductividad eléctrica de soluciones CIK a 25 °C.
| Concentración M |
Conductividad eléctrica μnho/cm |
|---|---|
| 10‒4 | 14,94 |
| 5 x 10‒4 | 73,90 |
| 10‒3 | 147 |
| 5 x 10‒3 | 717 |
| 10‒2 | 1.413 |
| 2 x 10‒2 | 2.767 |
| 5 x 10‒2 | 6.668 |
| 10‒1 | 12.900 |
| 2 x 10‒1 | 24.820 |
3.4 Procedimiento.
Dejar que la solución patrón de ClK y las muestras de agua estén a la misma temperatura. Cualquier temperatura entre 20 y 30 °C es aceptable, pero es importante que las soluciones problema y la patrón estén a la misma temperatura. Cuando se requiere gran precisión se colocarán las soluciones a 25 °C en termostato.
A continuación se enjuaga y se llena sucesivamente la celda de conductividad con la solución patrón y las soluciones problema y leyendo en el aparato (2-1), de acuerdo con su manual de instrucciones, los valores obtenidos.
3.5 Cálculos.

Siendo:
CE (x 106) = conductividad eléctrica.
CEClK = CE (x 106) de la solución patrón de ClK empleada.
R = resistencia en ohmios de la celda con la solución patrón.
R = resistencia en ohmios de la celda con la solución problema.
f = factor de corrección de temperatura de la tabla 3.1.
3.6 Observaciones.
3.6.1 Las células nuevas deben limpiarse con mezcla sulfocrómica, debiendo platinarse los electrodos de las mismas cuando los resultados sean erráticos o cuando se observe que el platino se ha desprendido total o parcialmente. Para ello se prepara una solución de 1 g de cloruro de platino y 0,012 g de acetato de plomo en 100 ml de agua. Se llena la celda con esta solución y se conectan los terminales de los electrodos a una batería de 1,5 voltios, intercalando un shunt en la batería cuando sea necesario ajustar la corriente, de manera que se desprenda una pequeña cantidad de gas. Dar por terminado el proceso cuando los electrodos estén recubiertos con un depósito de negro de platino. Drenar la célula y recuperar para un futuro uso la solución platinizante. Enjuagar bien la celda con agua destilada y dejarla llena de agua destilada cuando no se use.
3.6.2 Hasta 5.000 microohmios existe una aceptable correlación entre conductividad eléctrica y contenido salino de las aguas, de tal modo que los siguientes factores de conversión se manejan frecuentemente:


3.7 Referencias.
1. Bower, C. A., y Wilcox, L. V.: Methods of Soil Analysis Partz, pp. 937-940, 1965.
2. National Research Council: Inter. Critical tablas, 1929.
3. Agricultural Handbock, núm. 60. USDA, pp. 137-140, 1954.
4. Métodos de análisis recomendados por el Instituto de Hidrología del C. S. I. C., páginas 30-34, 1968.
TABLA 3.I
Factores de temperatura para corregir los datos de resistencia y conductividad de soluciones acuosas a la temperatura de 25 °C
CE25 = CEt x ft; R25 = Rt/ft
| 0 ºC | ft |
|---|---|
| 15,0 | 1,247 |
| 16,0 | 1,218 |
| 17,0 | 1,189 |
| 18,0 | 1,163 |
| 18,2 | 1,157 |
| 18,4 | 1,152 |
| 18,6 | 1,147 |
| 18,8 | 1,142 |
| 19,0 | 1,136 |
| 19,2 | 1,131 |
| 19,4 | 1,127 |
| 19,6 | 1,122 |
| 19,8 | 1,117 |
| 20,0 | 1,112 |
| 20,2 | 1,107 |
| 20,4 | 1,102 |
| 20,6 | 1,097 |
| 20,8 | 1,092 |
| 21,0 | 1,087 |
| 21,2 | 1,082 |
| 21,4 | 1,078 |
| 21,6 | 1,073 |
| 21,8 | 1,068 |
| 22,0 | 1,064 |
| 22,2 | 1,060 |
| 22,4 | 1,055 |
| 22,6 | 1,051 |
| 22,8 | 1,047 |
| 23,0 | 1,043 |
| 23,2 | 1,038 |
| 23,4 | 1,034 |
| 23,6 | 1,029 |
| 23,8 | 1,025 |
| 24,0 | 1,020 |
| 24,2 | 1,016 |
| 24,4 | 1,012 |
| 24,6 | 1,008 |
| 24,8 | 1,004 |
| 25,0 | 1,000 |
| 25,2 | 0,996 |
| 25,4 | 0,992 |
| 25,6 | 0,988 |
| 25,8 | 0,983 |
| 26,0 | 0,979 |
| 26,2 | 0,975 |
| 26,4 | 0,971 |
| 26,6 | 0,967 |
| 26,8 | 0,964 |
| 27,0 | 0,960 |
| 27,2 | 0,956 |
| 27,4 | 0,953 |
| 27,6 | 0,950 |
| 27,8 | 0,947 |
| 28,0 | 0,943 |
| 28,2 | 0,940 |
| 28,4 | 0,936 |
| 28,6 | 0,932 |
| 28,8 | 0,929 |
| 29,0 | 0,925 |
| 29,2 | 0,921 |
| 29,4 | 0,918 |
| 29,6 | 0,914 |
| 29,8 | 0,911 |
| 30,0 | 0,907 |
| 30,2 | 0,904 |
| 30,4 | 0,901 |
| 30,6 | 0,897 |
| 30,8 | 0,894 |
| 31,0 | 0,890 |
| 31,2 | 0,887 |
| 31,4 | 0,884 |
| 31,6 | 0,880 |
| 31,8 | 0,877 |
| 32,0 | 0,873 |
| 32,2 | 0,870 |
| 32,4 | 0,867 |
| 32,6 | 0,864 |
| 32,8 | 0,861 |
| 33,0 | 0,858 |
| 34,0 | 0,843 |
| 35,0 | 0,829 |
4. SULFATOS
(Determinación turbidométrica)
4.1 Principio.
El ion SO4= se precipita con ion Ba2+, en condiciones tales que se formen cristales de tamaño uniforme de SO4Ba, los que deben mantenerse en suspensión homogénea durante un periodo de tiempo que resulte suficiente para medir la absorbancia que la misma produzca.
El contenido en SO4= de cada muestra se obtiene a partir de la curva de calibrado previamente obtenida.
4.2 Material y aparatos.
4.2.1 Instrumento de medida, que puede ser uno de los siguientes.
4.2.1.1 Nefelómetro o turbidímetro.
4.2.1.2 Espectrofotómetro con medida a 420-430 nm.
4.2.1.3 Fotómetro de filtro, equipado con filtro de transmitancia 420-430 nm.
4.2.2 Tubos cilíndricos, calibrados a 50 cm3, con tapón.
4.3 Reactivos.
4.3.1 Solución de SO4K2 conteniendo 480 mg (10 meq) de SO4= por litro (0,8707 gramos de SO4K2 disueltos en agua destilada hasta completar un litro).
4.3.2 Solución precipitante de bario.‒Se disuelven 20 gramos de acetato bárico en una mezcla de 75 cm3 de ácido acético 10 N y 25 cm3 de solución de goma arábiga al 5 por 100, filtrando la solución resultante. El pH amortiguado de esta solución es de 3,70.
4.4 Procedimiento.
4.4.1 Obtención de la curva del calibrado.‒En los tubos cilíndricos 4.2.2 se introducen partes alícuotas de la solución 4.3.1. Se les añade agua destilada hasta unos 40 cm3, 1 cc de reactivo 4.3.2 y se completa a 50 cm3 con agua destilada. Se homogeneiza el conjunto durante un minuto por agitación suave y se deja en reposo al menos otro minuto. A partir de ese momento, y dentro de los quince minutos siguientes, pueden efectuarse las medidas; para ello, se traslada parte de la suspensión a las cubetas del aparato de medida que se utilice, midiendo las correspondientes absorbancias, empleando como blanco agua destilada sometida al mismo tratamiento.
Con los valores obtenidos se construye la curva de calibrado, que se aconseja tabular.
4.4.2 Valoración de las muestras.‒Se opera exactamente igual a como se ha indicado en 4.4.1, empleando 5 ml del problema.
4.5 Cálculo de los resultados.
El contenido en SO4= de las muestras se obtiene llevando las lecturas de absorbancias obtenidas con las mismas a la curva de calibrado 4.1 y teniendo en cuenta que se operó con 5 ml de muestra.
Los resultados pueden expresarse en meq o en mg de SO4= por litro.
4.6 Observaciones.
4.6.1 Si la lectura de una muestra sobrepasa la máxima concentración que figura en la curva de calibrado o inversamente de un valor muy bajo en dicha curva, debe repetirse el ensayo empleando menos de 5 ml o más de 5 ml de problema, respectivamente, lo que deberá tenerse en cuenta al efectuar el cálculo del resultado.
4.6.2 No se indican las concentraciones mínima y máxima, entre las que este método resulta válido, porque ello depende en gran manera del instrumento de medida que se utilice y del paso de luz de la cubeta empleada.
4.6.3 En esta técnica interfieren fundamentalmente el color y la turbidez. Esta puede eliminarse por filtración o centrifugación. La interferencia del color puede soslayarse utilizando la muestra coloreada como testigo, al que no se le agrega reactivo 4.3.2 o empleando como instrumento de medida un nefelómetro de doble posición de cubeta, con lo que se elimina la influencia del color.
4.7 Referencias.
1. Método utilizado en el Laboratorio de Suelos y Aguas del IRYDA. Inf. Quím. Analítica. V. pp. 1-6, 1951.
2. Métodos de análisis recomendados por el Instituto de Hidrología del Consejo Superior de Investigaciones Científicas, pp. 11-13, 1968.
5(a). CLORUROS
(Indicador)
5(a).1 Principio.
Los procedimientos clásicos para determinación de cloruros se basan en la formación de una sal de plata relativamente insoluble.
El punto de viraje de la valoración de cloruros con nitrato de plata puede ser detectado de diversas maneras, tal como por la aparición de un precipitado rojo de CrO4Ag2 (valoración de Mohr) o midiendo el potencial que se desarrolla en la solución mediante una combinación apropiada de electrodos (Kolthoff y Kuroda, 1951).
5(a).2 Material y aparatos.
5(a).2.1 Microbureta de 10 ml.
5(a).3 Reactivos.
5(a).3.1 Solución al 5 por 100 de cromato potásico.‒Disolver 5 g de cromato potásico en 50 ml de agua y añadir nitrato de plata 1 N, gota a gota, hasta que se forme un precipitado rojo claro permanente. Filtrar y diluir hasta 100 ml.
5(a).3.2 Nitrato de plata, 0,005 N.‒Disolver 0,8495 g de secado en estufa de nitrato de plata en agua y diluir exactamente en un litro. Almacenar la solución en un frasco color topacio para protegerla de la luz.
5(a).4 Procedimiento.
Tomar una alícuota de la solución problema y valorar con ácido sulfúrico hasta el punto de viraje con naranja de metilo (Métodos Oficiales de Análisis de Aguas, núm. 6). Se usará una solución patrón del ácido en caso de que interese determinar carbonatos y bicarbonatos.
Añadir cuatro gotas de reactivo 5(a).3.1 a la muestra guardada después de la determinación de carbonatos-bicarbonatos. Valorar, agitando al mismo tiempo bajo luz brillante, con el reactivo 5(a).3.2, usando una microbureta de 10 ml hasta que aparezca el primer color rojo (pardo rojizo) permanente. Las correcciones del ensayo en blanco varían con el volumen de la muestra en el punto de viraje, y generalmente aumenta regularmente entre 0,03 a 0,20 ml, según aumenta el volumen de 2 a 12 ml.
5(a).5 Cálculos.
Calcular la concentración de cloruros en meq/l.

Siendo:
V = Volumen, en ml, de NO3Ag utilizados en la valoración de la solución problema.
V' = Volumen, en ml, de NO3Ag utilizados en la valoración en blanco.
V'' = Volumen, en ml, de alícuota de solución problema.
5(a).6 Observaciones.
Los ácidos minerales que disuelven los cromatos de plata deben estar ausentes. Por tanto, la cantidad de ácido añadida debe ser la estrictamente necesaria para hacer la solución débilmente ácida, evitándose la adición en exceso. Los ioduros, bromuros y bicarbonatos forman precipitados con nitrato de plata y deben estar ausentes de la muestra. Generalmente, los fosfatos no interfieren, pero también deben de evitarse cantidades altas de fosfato. Los materiales orgánicos también deben de eliminarse por su tendencia a reducir el nitrato de plata en soluciones neutras. El hierro, normalmente, reacciona con el cromato potásico, produciendo un cromato insoluble, pero la cantidad de hierro que generalmente se encuentra en las muestras no interfiere. En caso de que se crea que la cantidad de hierro es alta, añadir unas cuantas gotas más de indicador para asegurar el exceso de cromato.
5(a).7 Referencias.
1. Champman, H. D., y Pratt, P. F.: Methods of Analysis for Soils Plants and Water, pp. 98-100, 1961.
2. Reitmeier, R. F.: Semimicroanalysis of Saline oil Solutions. Inds. and Engin. Chem. Analyt. Ed. 15, pp. 393-402, 1943.
3. Stout, P. R., y Johnson, C. M.: Methods of Soil Analysis. Part 2, pp. 1125-1126. American Society of Agronomy, 1965.
5(b). CLORUROS
(Potenciometría)
5(b).1 Principio.
Ver 5(a).1.
5(b).2 Material y aparatos.
5(b).2.1 Potenciómetro (pH-metro).
5(b).2.2 Electrodos de vidrio.
5(b).2.3 Electrodo de plata-cloruro de plata (Ag-ClAg).
5(b).2.4 Microbureta de 5 o 10 ml.
5(b).2.5 Agitador magnético.
5(b).3 Reactivos.
5(b).3.1 Suspensión de cloruro de plata (ClAg). Preparar un precipitado de ClAg mezclando soluciones 0,1 N de ClNa y NO3Ag; 500 ml de cada solución serán suficientes para precipitar ClAg para muchas determinaciones. Debe usarse un ligero exceso de cloruro o de nitrato para provocar un buen precipitado. Lavar bien el precipitado con agua destilada y transferirlo a un frasco topacio. Renovar la solución sobrenadante diariamente durante diez días, reemplazándola con agua destilada para remover las trazas de los excesos de iones plata o cloruro. Finalmente, completar el volumen con agua hasta un litro.
5(b).3.2 Solución de nitrato de plata (NO3Ag) 0,1 N. Disolver 8,5 g de NO3Ag en agua destilada, diluir la solución en un volumen de 500 ml y almacenar en un frasco topacio.
5(b).3.3 Solución de nitrato de plata (NO3Ag) 0,01 N. Diluir 10 ml de reactivo 5(b).3.2 en 100 ml con agua destilada. Comprobar antes de usarla su normalidad valorándola potenciométricamente con la solución CIK [reactivo 5(b).3.4], según se describe más adelante.
5(b).3.4 Solución patrón de cloruro potásico (CIK) 0,01 N. Disolver 0,746 g de CIK puro y desecado en estufa y diluir hasta un litro. Esta solución es 0,01 N con respecto al cloruro y se usa para valorar la solución NO3Ag.
5(b).3.5 Solución soporte de electrólito. Disolver 101 g de nitrato potásico (NO3K) recristalizado (calidad reactivo) en agua, añadir 62 ml de ácido nítrico concentrado (NO3H) y diluir la solución en un litro.
5(b).4 Procedimiento.
Transferir una alícuota de la solución problema preferentemente que contenga menos de 0,1 meq de cloruro al vaso de valoración. Diluir en agua destilada hasta un total de 25 ml, aproximadamente. Añadir 2,5 ml de la solución de electrólito.
Preparar la solución de referencia añadiendo a 25 ml de agua destilada 2,5 ml de la solución soporte de electrólito y dos gotas de la suspensión de ClAg. Sumergir el juego de electrodos de referencia, agitando ligeramente, cerrar el circuito del pH-metro y anotar el potencial observado en milivoltios. Reemplazar la solución de referencia por la solución problema que contenga la solución soporte de electrólito. Valorar agitando suavemente con NO3Ag 0,01 N hasta que el potencial que indique la escala del potenciómetro o pH-metro sea el mismo que el anotado para la solución de referencia. Anotar el volumen de solución de NO3Ag requerido.
5(b).5 Cálculo.
Calcular la concentración de Cl‒ en la solución problema, expresada en meq/l.

V = Volumen, en ml, de la solución de NO3Ag.
N = Normalidad de la solución de NO3Ag.
V' = Volumen, en ml, de la solución problema.
5(b).6 Observaciones.
5(b).6.1 Los vasos de valoración no deben exponerse a la luz solar directa durante la valoración, lo que podría provocar un cambio de potencial final. La agitación excesivamente rápida también puede cambiar el potencial final añadiendo una componente de potencial de corriente.
5(b).6.2 El uso de la solución soporte de electrólito de alta fuerza iónica dispensa de conocer al verdadero potencial de equivalencia al mismo tiempo que ahorra las correcciones necesarias por diferencia de fuerza iónica de la solución problema o clase de electrólitos disueltos. El «potencial aparente de equivalencia que se obtiene es prácticamente independiente de las variaciones normales al diferente contenido de sales solubles de la solución problema.
5(b).6.3 El procedimiento descrito no diferencia entre los iones cloruros y bromuros.
5(b).6.4 Existen en el mercado diversos equipos de valoración de cloruros en los que la valoración queda automáticamente interrumpida cuando se produce un cambio brusco de potencial o conductividad que equivale a la precipitación de todos los iones Cl‒ inicialmente presentes como ClAg.
Simultáneamente se expresa digitalmente la concentración de cloruros en la alícuota añadida a la solución soporte, en la que se realiza la valoración.
5(b).7 Referencias.
1. Fisher, R. B., y Peter, D. G.: Quantitatives Chemical analysis. Saunders, 1968.
2. Golterman, H. L.: Methods for Chemical analysis of fresh waters. TBP Hb60. Blackwell Oxford, 1970.
3. Kolthoff, I. M., y Kurola, P. K.: Determination of traces of chloride. Anal. Chem. 23: pp. 1034-1306, 1951.
4. Stout, P. R., y Johnson, C. M.: Methods of Soils Analysis. Pat 2, pp. 1127-1129. American Society of Agronomy, 1965.
6. ALCALINIDAD
6.1 Principio.
Se determina por valoración con una solución valorada en un ácido mineral fuerte a los puntos de equivalencia del bicarbonato (pH 8,3) y ácido carbónico (entre pH 4,2 y 5,4), bien sea potenciométricamente o por medio de indicadores. El segundo punto de equivalencia indica la alcalinidad total de la muestra, que puede ser debida a los siguientes iones: hidróxido, carbonatos y bicarbonatos. El pH al que se produce dicha equivalencia depende de la cantidad de CO2 en solución. Para una muestra dada la eliminación del CO2 en solución cuando la valoración se realice en atmósfera de nitrógeno o cuando se hierve en las proximidades del punto de viraje para suprimir el CO2, produce un cambio más brusco del potencial o del color, pero la posición del punto de viraje no cambia.
Suponiendo que las cantidades presentes en el agua de SiO4H3, BO3H2, NH4, SH‒ y CO3Ca coloidal o en suspensión sean despreciables, con las valoraciones mencionadas se pueden determinar las concentraciones individuales de hidróxidos, carbonatos y bicarbonatos.
6.2 Material y aparatos.
6.2.1 Matraces Erlenmeyer de 200 ml.
6.2.2 Pipeta.
6.2.3 Bureta.
6.2.4 Potenciómetro.
6.3 Reactivos.
6.3.1 Ácido sulfúrico o clorhídrico 0,1 N o 0,05 N, según la alcalinidad prevista de la muestra.
6.3.2 Indicador de fenolftaleína: Disolver 0,5 g de fenolftaleína en 50 ml de etanol de 95 por 100 y añadir 50 ml de agua. Añadir gota a gota solución de NaOH 0,05 N libre de CO2 hasta que el color se vuelva débilmente rosado.
6.3.3 Indicador de anaranjado de metilo al 0,05 por 100 de agua.
6.4 Procedimiento.
Tomar una alícuota que contenga menos de 1 meq de alcalinidad total y diluir hasta 50 ml con agua si fuese necesario. Añadir dos gotas de 6.3.2 si se produce color; valorar con 6.3.1 hasta que desaparezca el mismo. Añadir dos gotas de 6.3.3 y continuar la valoración con 6.3.2 hasta el punto de viraje del anaranjado de metilo. Caso de que exista dificultad en apreciar el viraje, detener la valoración en las proximidades del mismo, hervir el matraz durante unos minutos, dejar enfriar y continuar la valoración.
Hacer un ensayo en blanco con los reactivos y 50 ml de agua y restar de los anteriores valores los encontrados en este ensayo, caso de que fuesen apreciables.
En caso de utilizar potenciómetro, la primera valoración de la muestra se hará hasta pH = 3,8 y la segunda hasta pH = 4,0.
6.5 Cálculos.
a = ml de alícuota de la solución problema.
x = ml la lectura de la bureta en el punto de viraje de la fenolftaleína después de corregir por ensayo en blanco.
z = ml la lectura de la bureta en el punto de viraje del anaranjado de metilo después de corregir por ensayo en blanco.
N = normalidad del ácido.
Caso 1.° x < ½ z.
(OH‒) = despreciable


Caso 2.° x > ½ z.


(CO3H‒) = despreciable
Caso 3.° x = ½ z.
Concentraciones meq/l
(OH‒) y (CO3H‒) despreciables, aplicar para (SO3=) cualquiera de las dos igualdades de los casos 1.° y 2.°
Todos los casos.

6.6 Observaciones.
Todos los reactivos, así como el agua usada en diluciones y ensayos en blanco, deben obtener un bajo contenido en CO2, particularmente cuando se trate de muestras con alcalinidad baja. La eliminación del CO2 disuelto en el agua puede conseguirse mediante cualquiera de los dos procedimientos siguientes:
1) Reducir la presión durante diez a quince minutos con una trompa de agua.
2) Hervir durante diez a quince minutos y dejar enfriar en un Erlenmeyer con tapón.
6.7 Referencia.
1. Golterman, H. L.: Methods for Chemical analysis of fresh waters. IBP Handbook no. 8, Black Well Scientif. Publications. Oxford, 1970.
7. CALCIO
7.1 Principio.
Determinación directa por absorción atómica.
7.2 Material y aparatos.
7.2.1 Espectrofotómetro de absorción atómica.
7.2.2 Lámpara para determinar calcio.
7.2.3 Matraces aforados de 50, 100 y 1.000 ml de capacidad.
7.3 Reactivos.
7.3.1 Solución patrón de Ca de 0,05 N.
Disolver 2,500 g de CO3Ca en 30 cc de ClH (1:3), que se añade lentamente. Completar hasta un litro con agua destilada.
7.3.2 Solución de lantano al 6,5 por 100.
Disolver 76,22 g de óxido de lantano en 300 cc de ClH (1:1); añadir lentamente y con cuidado. Completar hasta un litro con agua destilada.
7.4 Procedimiento.
7.4.1 Obtención de la curva de calibrado.
Preparar a partir de 7.3.1 soluciones de Ca conteniendo 1,0 meq, 0,5 meq, 0,2 meq y 0,1 meq. A cada una de ellas se les adiciona con 40 cc de 7.3.2. Medir a continuación, en el espectrofotómetro de absorción atómica, las soluciones de calibrado a 422,7 nm utilizando llama de aire-acetileno. Con las lecturas obtenidas se traza la correspondiente curva de calibrado.
7.4.2 Valoración.
Introducir en un matraz aforado de 50 ml, 25 ml de la muestra y añadir 2 ml de solución de lantano 7.3.2. Completar a 50 ml con agua destilada. Continuar como en 7.4.1.
7.5 Cálculos.
Expresar los resultados indistintamente en meq o en mg de Ca por litro de agua los que se obtienen a partir de la curva de calibrado, teniendo en cuenta la alícuota de muestra utilizada.
8. MAGNESIO
8.1 Principio.
Determinación directa por absorción atómica.
8.2 Material y aparatos.
8.2.1 Espectrofotómetro de absorción atómica.
8.2.2 Lámpara para determinar magnesio.
8.2.3 Matraces aforados de 50, 100 y 1.000 ml de capacidad.
8.3 Reactivos.
8.3.1 Solución patrón de magnesio 0,05 N.
Atacar 0,600 g de metal puro con 30 cc de ClH (1:3), que se añade lentamente y calentando con suavidad. Una vez disuelto y frío el conjunto, completar hasta un litro con agua destilada.
8.3.2 Solución de lantano al 6,5 por 100.
Disolver 76,22 g de óxido de lantano en 300 cc de ClH (1:1); añadir lentamente y con cuidado. Completar hasta un litro con agua destilada.
8.4 Procedimiento.
8.4.1 Obtención de la curva de calibrado.
Preparar a partir de 8.3.1 soluciones de magnesio conteniendo 1,0 meq, 0,5 meq, 0,2 meq y 0,1 meq. A cada una de ellas se les adiciona con 40 cc de la 8.3.2. Medir a continuación, en el espectrofotómetro de absorción atómica, las soluciones de calibrado y 285,2 nm utilizando llama de aire acetileno. Con las lecturas obtenidas se traza la correspondiente curva patrón.
8.4.2 Valoración.
Introducir en un matraz aforado de 50 ml, 25 ml de la muestra y añadir 2 ml de solución de lantano 8.3.2. Completar a 50 ml con agua destilada. Continuar como en 8.4.1.
8.5 Cálculos.
Expresar los resultados indistintamente en meq o en mg de agua, los que se obtienen a partir de la curva de calibrado, teniendo en cuenta la alícuota de muestra utilizada.
9(a). NITRATOS
(Método colorimétrico)
9(a).1 Principio.
Reacción de los nitratos con la brucina en medio sulfúrico y posterior medida del color producido.
9(a).2 Material y aparatos.
9(a).2.1 Espectrofotómetro o colorímetro aptos para lecturas entre 400 y 425 nm.
9(a).2.2 Pipeta de seguridad.
9(a).3 Reactivos.
9(a).3.1 Solución patrón de nitrato. Disolver 0,1629 g de nitrato potásico anhidro en agua destilada y diluir a un litro. 1 ml de esta solución contiene 0,1 mg de NO3‒.
9(a).3.2 Reactivo brucina-ácido sulfanílico. Disolver 1,0 g de brucina y 0,1 g de ácido sulfanílico en unos 70 ml de agua destilada caliente, añadir 3 ml de ácido clorhídrico concentrado, dejar enfriar y diluir hasta 100 ml con agua destilada.
9(a).3.3 Solución de ácido sulfúrico. Agregar con cuidado 500 ml de ácido sulfúrico concentrado a 75 ml de agua destilada.
9(a).4 Procedimiento.
9(a).4.1 Construcción de la curva patrón. Tomar partes alícuotas de 9(a).3.1 y someterlas al procedimiento descrito en 9(a).4.2.
9(a).4.2 Determinación.
Introducir en un matraz aforado de 50 ml, 2,0 ml de la muestra. Añadir, mediante pipeta de seguridad, 1,0 ml de 9(a).3.2, y a continuación 10 ml de 9(a).3.3. Mezclar bien y dejar en la oscuridad 10 ± 1 minutos. Pasado este tiempo, añadir, agitando, agua destilada hasta 40 ml y dejar en la oscuridad durante 15 ± 1 minutos. A continuación colocar el matraz en un vaso con agua, dejándolo en la oscuridad hasta que adquiera la temperatura ambiente (generalmente quince minutos). Acto seguido enrasar el matraz con agua destilada, homogeneizar el conjunto, llevar al fotómetro y leer a 410 nm en cubeta de 1 cm de paso de luz. El valor 100 de transmitancia se obtiene con 2 ml de agua destilada sometida al mismo procedimiento indicado.
9(a).5 Cálculos.
Calcular el contenido en nitratos mediante comparación con la correspondiente curva patrón, teniendo en cuenta el factor de dilución.
9(a).6 Observaciones.
9(a).6.1 En las condiciones citadas se pueden medir concentraciones de NO3‒ comprendidas entre 5 y 50 mg de NO3‒ por litro. Si se desea medir concentraciones inferiores a la mínima citada, puede concentrarse la muestra o emplear cubetas de 2 o 4 cm de paso de luz. Inversamente, si de lo que se trata es de medir concentraciones superiores a la máxima que admite el procedimiento, se puede diluir la muestra o emplear cubetas de 0,5 cm de paso de luz.
9(a).6.2 Los iones Fe++, Fe+++ y Mn4+ pueden interferir ligeramente si su concentración es superior a 1 mg/l.
9(a).7 Referencias.
1. Standard Method, 14.a Ed., 1975, 427.
2. AOAC Method, 12.a Ed., 1975, 614.
9(b). NITRATOS
(Método por ultravioleta)
9(b).1 Principio.
Absorción de la radiación ultravioleta por el ion nitrato.
9(b).2 Material y aparatos.
9(b).2.1 Espectrofotómetro apto para lecturas a 220 y 275 nm.
9(b).2.2 Matraz aforado de 50 ml.
9(b).2.3 Pipeta graduada de 25 ml.
9(b).3 Reactivos.
9(b).3.1 Agua desionizada y destilada posteriormente en aparato de vidrio.‒Su empleo es necesario para la preparación de todas las soluciones y diluciones.
9(b).3.2 Solución patrón de nitrato.‒Disolver 0,7218 g de NO3K en agua y diluir a un litro. Esta solución contiene 100 mg de N por litro (solución A). Diluir 100 ml de la solución A a 1.000 ml con agua, 1 ml contiene 10 microgramos de N o 44,3 microgramos de NO3‒.
9(b).3.3 Solución de ácido clorhídrico 1 N.
9(b).3.4 Suspensión de hidróxido de aluminio.‒Disolver 125 g de alumbre de potasio en un litro de agua. Calentar a 60 °C y añadir 5 ml de NH3, lentamente y con agitación. Después de dejar la mezcla en reposo por espacio de una hora, llevarla a un vaso de dos litros y lavar el precipitado por sucesivas adiciones y decantaciones con agua, hasta eliminación de los iones cloruro, nitrato, nitrito y amonio. Finalmente, después de la sedimentación, decantar tanto líquido como sea posible, reservando solamente la suspensión concentrada.
9(b).4 Procedimiento.
9(b).4.1 Preparación de la muestra. Si se sospecha que la muestra de agua contiene interferencias orgánicas o es coloreada, añadir 4 ml de la suspensión de hidróxido alumínico a cada 100 ml de muestra en un Erlenmeyer. Agitar y dejar sedimentar durante cinco minutos. Pasar a través de un filtro de membrana de 0,45 micras previamente lavado con 200 ml de agua.
A 50 ml de la muestra clarificada como se expresa anteriormente o a 50 ml de la muestra filtrada por métodos convencionales. Añadir 1 ml de HCl 1 N y agitar.
9(b).4.2 Construcción de la curva patrón. Tomar partes alícuotas de 9(b).3.2 y someterlas al procedimiento descrito en 9(b) 4.3. La concentración estará comprendida entre 0 y 30 mg/l de NO3‒.
9(b).4.3 Determinación. Leer en el espectrofotómetro a 220 nm para obtener la lectura correspondiente a los nitratos y a 275 nm para obtener la interferencia debida a la materia orgánica disuelta. El valor cero de absorbancia o 100 de transmitancia se obtiene con agua 9(b).3.1 sometida al mismo procedimiento indicado.
9(b).5 Cálculos.
Restar la lectura a 275 nm multiplicada por dos de la lectura a 220 nm para obtener la absorbancia debida al ion nitrato.
Calcular el contenido en nitratos mediante comparación con la correspondiente curva patrón, teniendo en cuenta el factor de dilución.
9(b).6 Observaciones.
9(b).6.1 Interfieren la materia orgánica, carbonatos, nitritos, cromo hexavalente y detergentes aniónicos. Los carbonatos se eliminaron al añadir HCl; la materia orgánica, al restar su absorbancia de la del nitrato. Cuando se conocen las cantidades presentes en la muestra de nitritos, cromo hexavalente y detergentes aniónicos hay que construir las correspondientes curvas de corrección para cada una de estas sustancias por medida de sus absorbancias a 220 nm. El ion nitrito se puede también oxidar a nitrato con agua oxigenada de 110 volúmenes, teniéndolo en cuenta en los cálculos.
9(b).7 Referencia.
1. Standard Methods (APHA, AWWA y WPCF), 13.ª edición, 1971, 237.
10(a). AMONIO
(Método del reactivo de Nessler)
10(a).1 Principio.
El reactivo de Nessler (I2Hg 2 IK) produce con el amoníaco un complejo amarillo-pardo cuya intensidad de color puede medirse.
10(a).2 Material y aparatos.
10(a).2.1 Espectrofotómetro o fotocolorímetro apto para lecturas entre 400 y 425 nm.
10(a).2.2 Equipo de destilación con corriente de vapor de agua.
10(a).3 Reactivos.
10(a).3.1 Óxido de magnesio, exento de amoníaco.
10(a).3.2 Solución de ácido bórico al 2 por 100.
10(a).3.3 Reactivo Nessler. Disolver 100 g de I2Hg anhidro y 70 g de IK anhidro en el mínimo volumen de agua posible. Agregar esta disolución lentamente y agitando a una solución fría de 160 g de NaOH en 500 ml de agua. Diluir el conjunto hasta un litro con agua. Este reactivo debe dar el color característico con 0,1 mg/l de NH3 dentro de los diez minutos siguientes a su adición y no producir precipitado con 1 mg/l de NH3 en un espacio de tiempo de dos horas.
10(a).3.4 Solución madre de NH4+. Diluir 0,367 g de sulfato amónico en agua hasta un litro.
10(a).3.5 Solución patrón de NH4+. Diluir 100 ml de 10(a).3.4 con agua hasta un litro. 1 ml contiene 0,01 mg de NH4+.
10(a).4 Procedimiento.
10(a).4.1 Construcción de la curva patrón. Tomar partes alícuotas de 10(a).3.5 y someterlas al procedimiento descrito en 10(a).4.2, teniendo en cuenta la cantidad de muestra que se utilizó en la destilación y el volumen final del destilado.
10(a).4.2 Determinación.
Poner en marcha el equipo de destilación haciendo pasar por el mismo vapor de agua durante cinco minutos, al menos, para eliminar del equipo posibles trazas de amoníaco. Colocar a continuación en el matraz de destilación una cantidad exactamente medida de la muestra (250-300 ml), 0,5 g de MgO [10(a).3.1], poner en marcha el equipo recogiendo el destilado en un matraz Erlenmeyer de 100 ml, en el que se han colocado 5 ml de solución de ácido bórico [10(a).3.21. Cuando han destilado alrededor de 50 ml, comprobar si el destilado está ya libre de amoníaco, y si es así, suspender la operación, llevando el destilado a 100 ml con agua destilada en matraz aforado. Tomar 50 ml de la solución anterior, añadir 1 ml de reactivo Nessler [10(a).3.3] y mezclar el conjunto. Pasados diez minutos, llevar al espectrofotómetro donde se lee la absorbancia a 410-425 nm en cubeta de 1 cm de paso de luz. Como patrón cero utilizar 50 ml de agua sometidos al mismo tratamiento que la muestra.
10(a).5 Cálculos.
Calcular el contenido en amoníaco mediante comparación con la correspondiente curva patrón, teniendo en cuenta el factor de dilución.
10(a).6 Observaciones.
10(a).6.1 En las condiciones citadas se pueden medir concentraciones de amoníaco comprendidas entre 0,05 y 0,5 miligramos de NH4+ por litro. Si se desea medir concentraciones inferiores a la mínima citada, se pueden emplear cubetas de 2 a 4 cm de paso de luz. Si se trata de medir concentraciones superiores a la máxima que admite el procedimiento, diluir la muestra o emplear cubetas de 0,5 cm de paso de luz.
10(a).7 Referencias.
1. Standard Methods (APHA, AWWA, WPCF), 14.ª edición, 1975, 412.
2. AOAC Methods, 12.d edición, 1975, 614.
10(b). AMONIO
(Método volumétrico)
10(b).1 Principio.
El amoníaco destilado de la muestra se recoge sobre ácido bórico con un indicador adecuado y se valora con ácido sulfúrico de normalidad conocida. Aplicable a concentraciones superiores de 2 mg/l del ion amonio.
10(b).2 Material y aparatos.
10(b).2.1 Equipo de destilación con corriente de vapor de agua.
10(b).3 Reactivos.
10(b).3.1 Óxido de magnesio, exento de amoníaco.
10(b).3.2 Solución de ácido bórico al 2 por 100.
10(b).3.3 Indicador de Tshiro Tasiro. Disolver 0,125 g de rojo de metilo y 0,080 g de azul de metileno en 100 ml de alcohol de 96.
10(b).3.4 Ácido sulfúrico 0,005 N.
10(b).4 Procedimiento.
Poner en marcha el equipo de destilación haciendo pasar por el mismo vapor de agua durante cinco minutos al menos, para eliminar del equipo posibles trazas de amoníaco. Colocar a continuación en el matraz 250 ml de muestra, 1 g de MgO [10(b).3.1] y poner en marcha el equipo, recogiendo el destilado en un matraz Erlenmeyer de 250 ml en el que se han colocado 10 ml de solución de ácido bórico 10(b).3.2 y dos gotas del indicador 10(b).3.3. Comprobar, cuando han destilado alrededor de 50 ml, si el destilado está ya libre de amoníaco, y si es así, bajar el Erlenmeyer para que el pico del destilador quede por encima del líquido ya destilado; continuar la operación dos minutos más. Acto seguido, valorar el destilado con ácido sulfúrico 10(b).3.4. El indicador vira en el punto estequiométrico de verde a violeta.
10(b).5 Cálculos.
El contenido de la muestra, expresado en mg de NH4 por litro, se obtiene aplicando la siguiente expresión:
NH4+ (mg/l) = a. f. 0,09. 4
a = volumen, en ml, de ácido sulfúrico gastado en la valoración.
f = factor de corrección de la normalidad del ácido.
10(b).6 Referencias.
1. Standard Methods (APHA, AWWA, WPCF), 14.ª edición, 1975, 427.
2. AOAC Methods, 12.ª edición, 1975, 514.
11. NITRITOS
11.1 Principio.
Reacción con el ácido sulfanílico en medio clorhídrico y posterior medida espectrofotométrica del color desarrollado.
11.2 Material y aparatos.
11.2.1 Espectrofotómetro que permita lecturas a 425 nm.
11.3 Reactivos.
11.3.1 Reactivo de Zambelli. Diluir 260 ml de ácido clorhídrico concentrado con 500 ml de agua desionizada. Añadir 5,0 gramos de ácido sulfanílico y 7,5 g de fenol, calentando suavemente hasta disolución. Dejar enfriar y agregar 135 g de cloruro amónico. Cuando todo está disuelto, completar hasta 1 litro con agua desionizada
11.3.2 Amoníaco concentrado (d = 0,925).
11.3.3 Solución patrón de nitritos. Disolver 0,345 g de NO2Na en agua desionizada hasta 1 litro; 1 ml equivale a 0,230 mg de NO2‒. Esta solución se conserva hasta un mes añadiéndole 1 ml de cloroformo y manteniéndola en refrigerador. En caso contrario, deberá prepararse cada vez que se vaya a utilizar.
11.4 Procedimiento.
11.4.1 Curva patrón. Preparar las soluciones necesarias a partir de 11.3.3 mediante una o más diluciones llevadas a 50 ml con agua desionizada y sometidas a igual tratamiento de la muestra. Como blanco, usar 50 ml de agua desionizada sometidos a idéntico tratamiento que la muestra.
11.4.2 Tomar 50 ml de la muestra en un vaso y añadir mediante pipeta 2 ml del reactivo 11.3.1, mezclar bien y esperar durante diez minutos. A continuación añadir con pipeta 2 ml de 11.3.2. Homogeneizar el conjunto y esperar cinco minutos. Llevar la solución al espectrofotómetro y leer la absorbancia a 425 nm en cubeta de 1 cm de paso de luz. El color es estable veinticuatro horas.
11.5 Interpretación de resultados.
Partiendo de los valores de absorbancia obtenidos, hallar, mediante la curva patrón, la concentración de NO2‒ en la muestra.
12. SODIO
(Por fotometría de llama)
12.1 Principio.
Aspiración directa de la muestra a la llama y lectura a 586 nm.
12.2 Material y aparatos.
12.2.1 Espectrofotómetro o fotómetro de llama, aptos para efectuar lecturas a 586 nm.
12.3 Reactivos.
12.3.1 Solución de 1.000 mg Na/l. Pesar 2,542 g de cloruro sódico, previamente desecado, y disolver en agua desionizada, completando hasta 1 litro.
12.3.2 Soluciones patrones de 10, 20, 40, 60, 80 y 100 miligramos Na/l. Diluir partes alícuotas de la solución anterior a volumen conveniente.
12.3.3 Soluciones patrones de 1, 2, 4, 6, 8 y 10 mg Na/l. Diluir partes alícuotas de las soluciones anteriores a volumen conveniente.
12.4 Procedimiento.
Dejar estabilizar el aparato. Seleccionar la longitud de onda aspirando a la llama adecuada la solución patrón de 100 miligramos Na/l y recorrer cuidadosamente las proximidades de la longitud de onda teórica del sodio hasta encontrar aquella en que se logra la máxima respuesta.
Obtener las curvas de calibrado con la escala de patrones y a continuación aspirar la muestra y efectuar la lectura.
12.5 Expresión de los resultados.
En mg o meq de Na por litro de agua.
12.6 Observaciones.
12.6.1 Para reducir las posibles interferencias, añadir a la muestra y a las soluciones patrones litio u otro elemento fácilmente ionizable hasta una concentración del orden de 1.500 ppm.
12.6.2 Con determinado instrumental es necesario utilizar patrones más diluidos.
12.7 Referencias.
1. Standard Methods (APHA, AWWA, WPCF), 14.ª edición, 1975.
2. Instituto de Hidrología: «Normas Analíticas de las Aguas». Madrid, 1975.
13. POTASIO
(Por fotometría de llama)
13.1 Principio.
Aspiración directa de la muestra a la llama y lectura a 765 nm.
13.2 Material y aparatos.
13.2.1 Espectrofotómetro o fotómetro de llama, apto para efectuar lecturas a 765 nm.
13.3 Reactivos.
13.3.1 Solución de 1.000 mg K/l. Pesar 1,907 g de cloruro potásico, previamente desecado, y disolver en agua desionizada, completando hasta 1 litro.
13.3.2 Soluciones patrones de 10, 20, 40, 60, 80 y 100 miligramos K/l. Disolver partes alícuotas de la solución anterior a volumen conveniente.
13.3.3 Soluciones patrones de 1, 2, 4, 6, 8 y 10 mg K/l. Diluir partes alícuotas de las soluciones anteriores a volumen conveniente.
13.4 Procedimiento.
Dejar estabilizar el aparato. Seleccionar la longitud de onda aspirando a la llama adecuada la solución patrón de 100 miligramos K/l y recorrer cuidadosamente las proximidades de la longitud de onda teórica del potasio hasta encontrar aquella en que se logra la máxima respuesta.
Obtener las curvas de calibrado con la escala de patrones y a continuación aspirar la muestra y efectuar la lectura.
13.5 Expresión de los resultados.
En mg o meq de K por litro de agua.
13.6 Observaciones.
13.6.1 Para reducir las posibles interferencias, añadir a la muestra y a las soluciones patrones litio u otro elemento fácilmente ionizable hasta una concentración del orden de 1.500 ppm.
13.6.2 Con determinado instrumental es necesario utilizar patrones más diluidos.
13.7 Referencias.
1. Standard Methods (APHA, AWWA, WPCF), 14.ª edición, 1975.
2. Instituto de Hidrología: «Normas Analíticas de las Aguas». Madrid, 1975.
VINOS
42. INVESTIGACIÓN DE DERIVADOS MONOHALOGENADOS DEL ÁCIDO ACÉTICO
42.1 Principio.
Extracción de los derivados monohalogenados del ácido acético con éter sulfúrico previamente acidificado. Formación de tioíndigo, de color rojo, que se extrae con cloroformo.
El método permite detectar de 1,5 a 2 mg/l de ácido monocloroacético y las cantidades correspondientes de otros derivados monohalogenados.
42.2 Material y aparatos.
42.2.1 Éter sulfúrico.
42.2.2 Ácido sulfúrico (1 + 4) (v/v).
42.2.3 Sulfato sódico anhidro desecado en estufa a 105 °C.
42.2.4 Hidróxido sódico 0,5 N.
42.2.5 Ácido tiosalicílico (2 mercaptobenzoico) al 3 por 100 (p/v) en NaOH 1,5 N.
42.2.6 Ferricianuro potásico al 2 por 100 (preparar inmediatamente antes de usar).
42.2.7 Cloroformo.
42.2.8 Baño de agua.
42.2.9 Estufa eléctrica.
42.2.10 Embudos de separación.
42.2.11 Cápsulas de porcelana 70-80 mm ∅.
42.2.12 Tubos de 150 x 15 mm con tapón esmerilado.
42.2.13 Tubos de 150 x 15 mm con llave inferior.
42.3 Procedimiento.
Introducir en un Erlenmeyer de 500 cc y boca esmerilada 100 ml de vino, 5 ml de ácido sulfúrico (1 + 5) y, agitando, 100 ml de éter sulfúrico, continuando en un agitador durante una hora. Separar en embudo de decantación y descartar la capa interior. Añadir 8-10 g de sulfato sódico anhidro al extracto éter, agitando vigorosamente. Verter el éter en otro embudo de 500 ml, agregando sucesivamente: 5,0, 2,5 y 2,5 ml de NaOH 0,5 N, agitando un minuto cada vez y reuniendo las tres capas inferiores alcalinas obtenidas en un tubo con tapón esmerilado, en el cual se habrá vertido previamente 1 ml de la solución de ácido tiosalicílico. Agitar enérgicamente durante treinta segundos y transferir el contenido del tubo a una cápsula de porcelana, evaporando durante una hora en baño de agua (90-100 ºC), tiempo que habrá de respetarse aun cuando el agua se evapore mucho antes.
Si se formara una costra sobre la superficie, es conveniente romperla con una varilla fina de vidrio, lo que facilitará la evaporación. Pasado este tiempo, llevar la cápsula con el residuo seco a la estufa mantenida a 200 °C durante treinta minutos exactamente, pasados los cuales se retira y se deja enfriar a la temperatura ambiente. Añadir 2 ml de agua destilada que disolverá la mayor parte del extracto y recoger en un tubo con llave en su parte inferior; agregar otros 2 ml de agua destilada para lavar la cápsula y, por último, 3 ml de solución de ferricianuro, recogiendo todo ello con los primeros, agitando bien el tubo durante treinta segundos para facilitar la oxidación. Conviene asegurarse de que todo el residuo se disolvió bien en los 4 ml de agua destilada antes de añadir la solución de ferricianuro. En presencia de un derivado monohalogenado se desarrollará una coloración rojo-anaranjado (apenas perceptible para contenidos inferiores a 0,1 mg). Agregar 3 ml de cloroformo; agitar alternativamente tres-cuatro veces y decantar. Si el extracto clorofórmico inferior se colorea de rojo-fucsia o rosado, indica la presencia de derivados monohalogenados del ácido acético.
42.4 Referencias.
1. Recueil des Méthodes Internationales d’Analyse des Vins. O.l.V.A 35h.
2. Cabezuco, Gorostiza y Garrido: Determinación de derivados monohalogenados del ácido acético. Instituto de Fermentaciones (1971, ATA, vol. II, núm. 1, páginas 145-151).
43. GRADO ALCOHÓLICO EN POTENCIA
43.1 Principio.
Es el volumen en litros de alcohol susceptible de obtenerse por fermentación total de los azúcares contenidos en 100 litros de vino.
43.2 Material y aparatos.
Como en 7(a).2 o 7(b).2.
43.3 Reactivos.
Como en 7(a).3 o 7(b).3.
43.4 Procedimiento.
Como en 7(a) o 7(b).
43.5 Cálculos.

Donde:
N = g/l de azúcares reductores de la muestra.
44. GRADO ALCOHÓLICO TOTAL
44.1 Principio.
Se considera el grado alcohólico total como la suma del grado alcohólico adquirido determinado según 5(a) o 5(b), y del grado alcohólico potencial, determinado según 43.
44.2 Cálculo.
Grado alcohólico total = Grado alcohólico adquirido + Grado alcohólico potencial.
7(c). AZÚCARES REDUCTORES
(Valoración)
7(c).1 Principio.
Eliminación de las materias reductoras distintas de los azúcares reductores por defecación, y posterior valoración basada en la acción reductora de los azúcares sobre una solución cupro-alcalina.
7(c).2 Material y aparatos.
7(c).2.1 Erlenmeyer de 300 ml con refrigerante de reflujo.
7(c).2.2 Material necesario para volumetría.
7(c).2.3 Baño de agua.
7(c).3 Reactivos.
7(c).3.1 Solución cupro-alcalina:
Disolver por separado: 25 g de sulfato de cobre puro (SO4Cu • 5H2O) en 100 ml de agua: 50 g de ácido cítrico en 300 ml de agua y 388 g de carbonato de sodio cristalizado en 300-400 ml de agua caliente. Mezclar la solución de ácido cítrico y la de carbonato de sodio. Añadir a continuación la solución de sulfato de cobre y completar el volumen con agua hasta un litro.
7(c).3.2 Solución de ioduro de potasio al 30 por 100. Conservar en frasco topacio.
7(c).3.3 Solución de ácido sulfúrico al 25 por 100 en volumen.
7(c).3.4 Solución de engrudo de almidón de 5 g/l; contendrá 200 g/l de cloruro de sodio para asegurar su conservación. Esta solución debe de ser mantenida diez minutos en ebullición en el momento de su preparación.
7(c).3.5 Tiosulfato de sodio N/10.
7(c).4 Procedimiento.
Poner en el Erlenmeyer de 300 ml, 25 ml de la solución cuproalcalina y 25 ml del vino previamente defecado según 7(a) o 7(b). Este volumen añadido no debe contener más de 60 mg de azúcares reductores.
Añadir algunos granos de piedra pómez y llevar a ebullición, que debe de ser alcanzada en dos minutos, adaptando el Erlenmeyer al refrigerante de reflujo y mantener exactamente durante diez minutos la ebullición.
Enfriar inmediatamente bajo corriente de agua fría. Añadir 10 ml de la solución de ioduro de potasio al 30 por 100, 25 ml de la solución de ácido sulfúrico al 25 por 100 y 2 ml de engrudo de almidón. A continuación, valorar con la solución 0,1 N de tiosulfato de sodio.
Efectuar una prueba en blanco, sustituyendo los 25 ml del vino previamente defecados por igual volumen de agua destilada y tratar como se ha indicado antes para la muestra.
7(c).5 Cálculos.
La cantidad de azúcar, expresada en azúcar invertido contenida en la muestra analizada, se obtiene en la tabla 7(c).1 en función del n' ‒ n de ml de tiosulfato utilizado.
Siendo:
n = Volumen, en ml, de solución de tiosulfato de sodio 0,1 N utilizado en la valoración de la muestra.
n' = Volumen, en ml, de solución de tiosulfato de sodio 0,1 N utilizado en la prueba en blanco.
Expresar el contenido del vino en g de azúcar invertido por litro, teniendo en cuenta las diluciones efectuadas en el curso de la defecación del volumen de la muestra analizada.
7(c).6 Referencias.
1. Recueil des Méthodes Internationales d’Analyse des Vins. O.I.V. A 4e 29-31.
TABLA 7(c).1
Azúcares reductores, expresados en mg
| MI N/10 de tiosulfato de sodio | Primera cifra decimal | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
| 0 | 0,0 | 0,3 | 0,6 | 1,0 | 1,3 | 1,6 | 1,9 | 2,2 | 2,6 | 2,9 |
| 1 | 3,2 | 3,5 | 3,8 | 4,2 | 4,5 | 4,8 | 5,1 | 5,4 | 5,7 | 6,1 |
| 2 | 6,4 | 6,7 | 7,1 | 7,4 | 7,7 | 8,1 | 8,4 | 8,7 | 9,0 | 9,4 |
| 3 | 9,7 | 10,0 | 10,4 | 10,7 | 11,0 | 11,4 | 11,7 | 12,0 | 12,3 | 12,7 |
| 4 | 13,0 | 13,3 | 13,7 | 14,0 | 14,4 | 14,7 | 15,0 | 15,4 | 15,7 | 16,1 |
| 5 | 16,4 | 16,7 | 17,1 | 17,4 | 17,8 | 18,1 | 18,4 | 18,8 | 19,1 | 19,5 |
| 6 | 19,8 | 20,1 | 20,5 | 20,8 | 21,2 | 21,5 | 21,8 | 22,2 | 22,5 | 22,9 |
| 7 | 23,2 | 23,5 | 23,9 | 24,2 | 24,6 | 24,9 | 25,2 | 25.6 | 25,9 | 26,3 |
| 8 | 26,6 | 26,9 | 27,3 | 27,6 | 28,0 | 28,3 | 28,6 | 29,0 | 29,3 | 29,7 |
| 9 | 30,0 | 30,3 | 30,7 | 31,0 | 31,3 | 31,7 | 32,0 | 32,4 | 32,7 | 33,0 |
| 10 | 33,4 | 33,7 | 34,1 | 34,4 | 34,8 | 35,1 | 35,4 | 35,8 | 36,1 | 36,5 |
| 11 | 36,8 | 37,2 | 37,5 | 37,8 | 39,2 | 38,6 | 38,9 | 39,3 | 39,6 | 40,0 |
| 12 | 40,3 | 40,7 | 41,0 | 41,4 | 41,7 | 42,1 | 42,4 | 42,8 | 43,1 | 43,5 |
| 13 | 43,8 | 44,2 | 44,5 | 44,9 | 45,2 | 45,6 | 45,9 | 46,3 | 46,6 | 47,0 |
| 14 | 47,3 | 47,7 | 48,0 | 48,4 | 48.7 | 49,1 | 49,4 | 49,8 | 50,1 | 50,5 |
| 15 | 50,8 | 51,2 | 51,5 | 51,9 | 52,2 | 52,6 | 52,9 | 53,3 | 53,6 | 54,0 |
| 16 | 54,3 | 54,7 | 55,0 | 55,4 | 55,8 | 56,2 | 56,5 | 56,9 | 57,3 | 57,6 |
| 17 | 58,0 | 58,4 | 58,8 | 59,1 | 59,5 | 59,9 | 60,3 | 60,7 | 61,0 | 61,4 |
| 18 | 61,8 | 62,2 | 62,5 | 62,9 | 63,3 | 63.7 | 64,0 | 64,4 | 64,8 | 65,1 |
| 19 | 65,5 | 65,9 | 66,3 | 66,7 | 67,1 | 67.5 | 67,8 | 68,2 | 68,6 | 69,0 |
| 20 | 69,4 | 69,8 | 70,2 | 70,6 | 71,0 | 71,4 | 71,7 | 72,1 | 72,5 | 72,9 |
| 21 | 73,3 | 73,7 | 74,1 | 74,5 | 74,9 | 75,3 | 75,6 | 76,0 | 76.4 | 76,8 |
| 22 | 77,2 | 77,6 | 78,0 | 78,4 | 78,8 | 79,2 | 79,6 | 80.0 | 80,4 | 80,8 |
| 23 | 81,2 | 81,6 | 82,0 | 82,4 | 82,8 | 83,2 | 83,6 | 84,0 | 84,4 | 84,8 |
| 24 | 85,2 | 85,6 | 86,0 | 86,4 | 86,8 | 87,2 | 87,6 | 88,0 | 88,4 | 88,8 |
| 25 | 89,2 | 89,6 | 90,0 | 90,4 | 90,8 | 91,2 | 91,6 | 92,0 | 92,4 | 92,8 |
30. CLORUROS
30.1 Principio.
Determinación directa de cloruros por potenciometría utilizando el electrodo Ag/AgCl.
30.2 Material y aparatos.
30.2.1 pH-metro con escala que permita apreciar 2 mV.
30.2.2 Agitador magnético.
30.2.3 Electrodo Ag/AgCl, con una solución saturada de nitrato de potasio como electrólito.
30.2.4 Microbureta graduada en 1/100 de ml.
30.3 Reactivos.
30.3.1 Solución patrón de cloruros.‒Disolver 2,1027 g de cloruro de potasio puro para análisis (máximo 0,005 por 100 de bromo previamente desecado, en agua destilada hasta 1 litro. 1 ml de esta solución contiene 1 mg de ion cloro.
30.3.2 Solución de nitrato de plata.‒Disolver 4,7912 g de nitrato de plata en una solución alcohólica al 10 por 100 (v/v) hasta 1 litro, 1 ml de esta solución corresponde a 1 mg de ion cloro.
30.3.3 Ácido nítrico concentrado (d = 1,40).
30.4 Procedimiento.
30.4.1 Determinación del potencial del punto de equivalencia.
Introducir 5,0 ml de la solución patrón de cloruros en un vaso de 150 ml. Diluir aproximadamente a 100 ml con agua destilada y acidificar con 10 ml de ácido nítrico (30.3.3). Introducir los electrodos. Añadir 10 ml de la solución de nitrato de plata, agitando moderadamente: adicionar los cuatro primeros mililitros en fracciones de 1 ml y leer las correspondientes lecturas en milivoltios; los dos siguientes mililitros, en fracciones de 0,2 ml y continuar adicionando en fracciones de 1 mililitro hasta que se hayan alcanzado un total de 10 ml. Después de cada adición esperar unos treinta segundos antes de hacer la correspondiente lectura en milivoltios. Llevar los valores así obtenidos sobre un papel milimetrado en función de los correspondientes mililitros de solución de nitrato de plata y a partir del punto singular de la curva obtenida determinar el potencial del punto de equivalencia.
Para comprobar el potencial del punto de equivalencia, llevar a un vaso de 150 ml 5,0 ml de la solución patrón de cloruros, 95 ml de agua destilada y 1 ml de ácido nítrico concentrado. Introducir el electrodo y valorar agitando, justamente hasta el potencial del punto de equivalencia.
Repetir esta operación hasta obtener una buena concordancia de resultados. Este control debe efectuarse antes de determinar la concentración de cloruros en cada serie de muestras a analizar.
30.4.2 Determinación.
Llevar a un vaso de 150 ml 50,0 ml de vino. Añadir 50 ml de agua destilada y 1,0 ml de ácido nítrico concentrado. Valorar a continuación siguiendo el procedimiento descrito anteriormente hasta alcanzar el potencial del punto de equivalencia.
30.5 Cálculos.
La cantidad de cloruros se determina mediante las expresiones:
0,02 n en gramos de ion cloro.
0,5633 n en miliequivalentes por litro de ion cloro.
0,0329 n en gramos de cloruro sódico por litro.
Siendo:
n = volumen, en ml, de solución de nitrato de plata necesario para alcanzar el potencial del punto de equivalencia.
30.6 Referencia.
1. Recueil des Méthodes Internationales d’Analyse des Vins. O. I. V. A 15j.
45. COLORANTES SINTÉTICOS
45.1 Principio.
Concentración mediante ebullición, extracción con éter y fijación de colorantes sintéticos básicos y ácidos sobre lana y posterior confirmación por cromatografía en papel.
45.2 Material y aparatos.
45.2.1 Hebras de lana blanca previamente desengrasadas con éter.
45.2.2 Hebras de lana blanca mordentadas. Disolver 1 g de sulfato de aluminio cristalizado y 1,2 g de tartrato ácido de potasio en 500 ml de agua. Introducir en esta solución 10 g de hebras de lana blanca, previamente desengrasadas con éter y secas, agitar durante una hora y dejar reposar de dos a tres horas; transcurrido dicho tiempo, secar a temperatura ambiente.
45.2.3 Papel para cromatografía Whatman número 1 o similar.
45.2.4 Erlenmeyer de 50 ml de capacidad.
45.2.5 Ampolla de decantación de 200 ml de capacidad.
45.3 Reactivos.
45.3.1 Éter sulfúrico.
45.3.2 Hidróxido de sodio al 5 por 100.
45.3.3 Ácido acético glacial, d = 1,05.
45.3.4 Ácido acético diluido, 1/18 (v/v).
45.3.5 Ácido clorhídrico diluido 1/10 (v/v).
45.3.6 Amoníaco puro, d = 0,92.
45.3.7 Solvente número 1 para cromatografía de colorantes de carácter básico:
Butanol n: 50 ml.
Etanol: 25 ml.
Ácido acético glacial: 10 ml.
Agua destilada: 25 ml.
45.3.8 Solvente número 2 para la cromatografía de colorantes de carácter ácido:
Butanol n: 50 ml.
Etanol: 25 ml.
Amoníaco puro: 10 ml
Agua destilada: 25 ml.
45.4 Procedimiento.
45.4.1 Determinación de colorantes de carácter básico.
Poner 200 ml de vino en un Erlenmeyer de 500 ml y llevar a ebullición hasta que el vino se reduzca a un tercio de su volumen. Enfriar y neutralizar con la solución de hidróxido sódico al 5 por 100 hasta claro viraje del color natural del vino. Extraer dos veces, con 30 ml de éter cada vez, y juntar las dos fases etéreas; en ellas se encuentran eventualmente los colorantes básicos; el residuo de la extracción debe ser conservado con objeto de investigar los colorantes ácidos. Lavar dos veces el éter empleado en la extracción con 5 ml de agua, con el objeto de eliminar el hidróxido sódico; a continuación, añadir 5 ml de ácido acético diluido a la fase etérea; la presencia de un colorante básico colorea la fase ácido-acuosa; la presencia de este colorante puede confirmarse por su fijación sobre lana mordentada.
La fase ácido-acuosa obtenida se alcaliniza con amoníaco al 5 por 100; añadir 0,5 g de lana mordentada y llevar a ebullición durante un minuto; aclarar la lana bajo el chorro de agua fría; de permanecer coloreada, el vino contiene un colorante básico.
45.4.1.1 Caracterización por cromatografía de papel. A partir de la fase acético-acuosa que contiene el colorante básico, concentrar hasta 0,5 ml. Si el colorante ha sido fijado sobre lana, añadir a ésta 10 ml de agua destilada, 10 gotas de ácido acético y llevar a ebullición, y una vez retirada la lana previamente escurrida, concentrar la solución hasta 0,5 ml.
Depositar sobre papel de cromatografía 20 microlitros de la anterior solución concentrada a 3 cm del borde lateral y a 2 cm del borde inferior. Introducir dicho papel en una cubeta que contenga el solvente 45.3.7 de forma que su borde inferior esté sumergido en el solvente 1 cm. Retirar y dejar secar al aire cuando se haya alcanzado una altura del frente del solvente comprendido entre 20 o 25 cm. Identificar el colorante por medio de soluciones de colorantes sintéticos básicos depositados simultáneamente sobre el cromatograma.
45.4.2 Determinación de colorantes de carácter ácido.
Partir del residuo de vino concentrado a un tercio y neutralizado después de su extracción con éter; si no se realizó la determinación de colorante de carácter básico, poner 200 ml de vino en un Erlenmeyer de 500 ml y llevar a ebullición hasta que el vino se reduzca a un tercio de su volumen. En uno u otro caso, añadir 3 ml de ácido clorhídrico diluido (45.3.5) y 0,5 g de lana blanca; hervir durante cinco minutos, decantar el líquido y lavar la lana con agua abundante. En el Erlenmeyer que contiene la lana, añadir 100 ml de agua y 2 ml de ácido clorhídrico diluido (45.3.5); hervir durante cinco minutos, decantar el líquido ácido y repetir esta operación hasta que el líquido de lavado sea incoloro. Después de haber lavado bien la lana, para eliminar completamente el líquido ácido, añadir 50 ml de agua destilada y 10 gotas de amoníaco puro; llevar a ebullición suave durante diez minutos a fin de disolver la materia colorante artificial que eventualmente se haya fijado sobre la lana. A continuación, retirar la lana del Erlenmeyer, llevar el volumen de líquido a 100 ml y hervir hasta evaporación completa del amoníaco; una vez conseguida dicha evaporación, añadir 2 ml de ácido clorhídrico diluido (45.3.5) (comprobar que el líquido haya adquirido reacción francamente ácida, llevando una gota de éste sobre el papel indicador). Poner en el Erlenmeyer 60 mg de lana blanca y llevar a ebullición durante cinco minutos; retirar la lana y aclararla bajo el chorro de agua fría. Si después de esta operación la lana toma coloración roja, cuando se trata de vino tinto, o amarilla, si se trata de vino blanco, confirma la presencia de materia colorante orgánica artificial de naturaleza ácida. Si la coloración adquirida es débil o dudosa, repetir el tratamiento con amoníaco y hacer una segunda fijación sobre una hebra de lana de 30 mg. Si transcurrida esta segunda fijación se obtiene una coloración rosa, aunque sea débil, se deduce la presencia de colorante ácido. Recurrir para un análisis más completo a nuevas fijaciones-eluciones (hasta 4 o 5), operando siempre de forma idéntica a la empleada para la segunda fijación hasta que se obtenga una coloración rosácea que, aunque sea pálida, no ofrece lugar a dudas.
45.4.2.1 Caracterización por cromatografía de papel.
Añadir sobre la hebra de lana coloreada 10 ml de agua destilada, lo gotas de amoníaco y llevar a ebullición; retirar la lana previamente escurrida y concentrar la solución amoniacal hasta 0,5 ml. Depositar sobre papel de cromatografía 20 microlitros de la anterior solución concentrada, a 3 cm del borde lateral y a 2 cm del borde inferior; introducir dicho papel en una cubeta que contenga el solvente 45.3.8 de forma que su borde inferior esté sumergido en el solvente l cm. Retirar y dejar secar al aire cuando se haya alcanzado una altura del frente del solvente comprendida entre 20 o 25 cm. Identificar el colorante por medio de soluciones de colorantes sintéticos ácidos depositados simultáneamente sobre el cromatograma.
45.5 Referencia.
Recueil des méthodes internationales d’Analyse des vins. A43k, páginas 1-6.
46. MERCURIO
46.1 Principio.
Oxidación de la materia orgánica con ácido nítrico y crómico, reducción de las sales mercúricas por acción de Sn++, volatilización del Hg metálico, arrastre del mismo por corriente de aire y determinación de su absorbancia a 253,7 nanómetros.
Este método es aplicable a concentraciones de mercurio superiores a 0,05 mg/l.
46.2 Material y aparatos.
46.2.1 Espectrofotómetro de absorción.
46.2.2 Lámpara de mercurio.
46.2.3 Aparato para la reducción del ion mercúrico a mercurio metal y posterior arrastre de éste con corriente de aire (figura 46.1).
46.2.4 Matraces de reacción de forma cónica de 100 ml de capacidad.
46.2.5 Bomba peristáltica accionada con motor provisto de vaciador de velocidad.
46.2.6 Llave de purga en T de teflón.
46.2.7 Tubo flexible de tigón de 1/8 pulgada de diámetro interior y un metro, aproximadamente, de longitud.
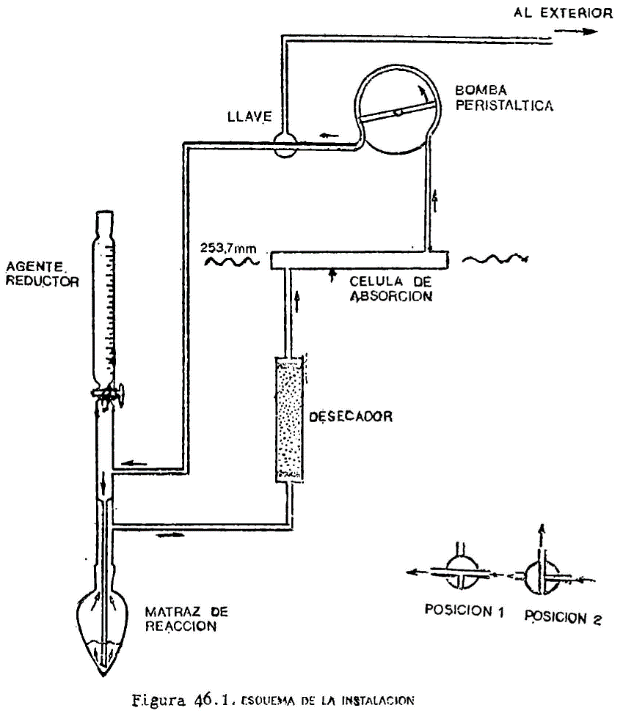
46.3 Reactivos.
46.3.1 Ácido sulfúrico, exento de mercurio.
46.3.2 Oxido de mercurio.
46.3.3 Cloruro estannoso, exento de mercurio.
46.3.4 Oxido de cromo, exento de mercurio.
46.3.5 Solución de ácido crómico al 5 por 100.‒Llevar a un vaso de 500 ml 25 g de óxido de cromo y disolverlos en 25 ml de agua destilada. Añadir lentamente y agitando ácido sulfúrico concentrado hasta enrasar a 500 ml cuando la solución esté fría.
46.3.6 Solución reductora.‒Solución al 40 por 100 de cloruro estannoso en agua destilada, acidulada con sulfúrico.
46.3.7 Solución patrón.‒Disolver 1,080 g de óxido de mercurio en 100 ml de ácido clorhídrico al 50 por 100; enrasar con agua a 1.000 ml. Mediante dilución 1/1.000 se obtiene la solución de 1 mg/l.
46.4 Procedimiento.
46.4.1 Preparación de la muestra.
Tomar 1 ml de muestra y colocarla dentro de un matraz cónico. Añadir 2 ml de ácido nítrico. Cerrar con película de parafina y dejarlo en digestión durante sesenta minutos a temperatura ambiente. Añadir 5 ml de ácido crómico al 5 por 100, refrigerando durante la adición en baño de hielo. Tapar el matraz y dejarlo durante sesenta minutos, agitando cada diez minutos. Transcurrido este tiempo, añadir 15 ml de agua destilada, enfriando durante la adición con baño de hielo. Una vez fríos los matraces, añadir 2 g, aproximadamente, de sulfato de hidroxilamina, agitar y acoplar los matraces al aparato de medida. Añadir 10 ml de solución reductora. Continuar como en 46.4.3.
46.4.2 Construcción de la curva patrón.
A partir de 46.3.7, tomar 0,5, 10, 15, 20 y 25 ml. Tratarlas a continuación como se indica en 46.4.1. Estas soluciones contienen 0, 0,1, 0,2, 0,3, 0,4 y 0,5 partes por millón de Hg. Anotar las absorbancias obtenidas frente a las concentraciones correspondientes.
46.4.3 Determinación.
Efectuar la lectura de la absorbancia de la muestra. Una vez preparada según 46.4.1.
46.5 Referencias.
1. A. Bouchard: «Determination of mercury after soon temperature digestión by flamaless atomic absorption.» Atom. Absorpt. Newsletter, vol. 12, número 5 (115-117), 1973.
2. A. García de Jalón y J. Frías: «Un método para la determinación de mercurio en vinos mediante espectrofotometría de absorción atómica sin llama.» Ministerio de Agricultura. Abril 1972.
47. PLOMO
47.1 Principio.
Determinación del plomo por A. A. después de una concentración previa con objeto de conseguir resultados suficientemente precisos.
47.2 Material y aparatos.
47.2.1 Espectrofotómetro de absorción atómica.
47.2.2 Lámpara de plomo.
47.2.3 Cápsula de platino de 100 ml de capacidad o similar.
47.2.4 Baño de arena.
47.2.5 Mufla con circulación de aire.
47.2.6 Matraces de 10, 100 y 1.000 ml de capacidad.
47.3 Reactivos.
47.3.1 Ácido sulfúrico del 96 por 100 (d = 1,835).
47.3.2 Ácido nítrico del 70 por 100 (d = 1,413).
47.3.3 Ácido nítrico al 1 por 100 en agua destilada (v/v).
47.3.4 Solución patrón de 1.000 ppm de plomo. Disolver 1,598 gramos de (NO3)2Pb enrasando a 1.000 ml con ácido nítrico al 1 por 100.
47.4 Procedimiento.
47.4.1 Preparación de la muestra.‒Poner 100 ml de la muestra en una cápsula de platino y llevarla a evaporación hasta consistencia siruposa en baño de arena. Añadir a continuación 2 ml de ácido sulfúrico y carbonizar el residuo en el baño de arena. Seguidamente introducir la cápsula en la mufla y mantenerla durante dos horas a 450 °C; transcurrido dicho tiempo, sacarla y dejarla enfriar. Añadir 1 ml de ácido nítrico concentrado, evaporar en el baño de arena e introducir en la mufla, repitiendo esta operación hasta obtener cenizas blancas. Disolver a continuación las cenizas con 1 ml de ácido nítrico concentrado y 2 ml de agua destilada, una vez disueltas filtrar y recoger el filtrado en un matraz de 10 ml, lavando la cápsula y el filtro con agua destilada hasta el enrase.
47.4.2 Construcción de la curva patrón.‒A partir de la solución patrón 46.3.4, tomar alícuotas de 0,2; 0,4; 0,6; 0,8, y 1 ml, y llevar a 100 ml con ácido nítrico al 1 por 100 (v/v).
El contenido en plomo de estas soluciones es, respectivamente, 2, 4, 6, 8 y 10 p.p.m. Anotar las absorbancias obtenidas frente a las concentraciones correspondientes.
47.4.3 Determinación.‒Efectuar la lectura directa de la muestra preparada según 47.4.1 a 283,3 nm.
47.5 Cálculos.
Calcular el contenido en plomo, expresado en p.p.m. mediante comparación con la correspondiente curva patrón y teniendo en cuenta e1 factor de dilución.
39(b). FLÚOR
39(b).1 Principio.
Determinación directa del flúor total por potenciometría utilizando un electrodo iónico específico.
Para eliminar las posibles causas de error se le adiciona al vino una solución tampón.
39(b).2 Material y aparatos.
39(b).2.1 pH-metro con escala que permita apreciar 0,1 mV.
39(b).2.2 Electrodo selectivo de flúor.
39(b).2.3 Electrodo de referencia de calomelanos.
39(b).2.4 Microbureta graduada en 1/100 de ml.
39(b).2.5 Recipientes de plástico de 50-100 ml de capacidad para mezclas y matraces en material de plástico para conservar las soluciones patrón.
39(b).2.6 Pipetas de precisión graduadas en microlitros.
39(b).2.7 Agitador magnético.
39(b).3 Reactivos.
39(b).3.1 Solución patrón de fluoruros 10‒2 M conteniendo 190 mg de F‒ por litro. Pesar 0,4198 g de FNa (desecado durante cuatro horas a 106 °C antes de su utilización), disolver y enrasar a un volumen de 1.000 mL
39(b).3.2 Solución tampón de PO4H3 0,75 M.
39(b).4 Procedimiento.
39(b).4.1 Verificación del electrodo y determinación de su pendiente.
A partir de la solución 3.1 preparar, mediante diluciones, soluciones de concentraciones conocidas de 10‒3 M, 10‒4 M y 10‒5 M, que contienen, respectivamente, 19, 1,9 y 0,19 mg de F‒, por litro.
Tomar dos vasos de precipitados; en uno de ellos colocar 25 ml de la disolución 10‒3 M de F‒, adicionar 5 ml de solución tampón de PO4H3 y determinar su potencial agitando de forma moderada mientras dure la medida. En el otro vaso, poner 25 ml de disolución 10‒4 M de F‒, 5 ml de solución tampón de PO4H3 y determinar su potencial, agitando como anteriormente. La diferencia entre los dos potenciales es la pendiente del electrodo (S) y cuyo valor debe ser, aproximadamente, 59 mV.
39(b).4.2 Determinación.
Tomar 25 ml de vino, adicionar 5 ml de la solución tampón y determinar su potencial (E1). A continuación, y en el mismo vaso, adicionar un ml de disolución 10‒3 M de F‒ y determinar su potencial (E2).
Si el salto de potencial es pequeño, indica que la concentración de F‒ es muy alta y hay que diluir la muestra hasta que el salto alcanzado esté comprendido alrededor de 20-40 mV.
Los volúmenes de muestra y solución tampón pueden ser modificados en función de la forma y disposición de los electrodos utilizados.
39(b).5 Cálculos.
La cantidad de flúor, expresada en mg/l, nos viene determinada por la fórmula:

Siendo:
CF‒ = concentración, en mg/l, de F‒ en el medio.
V = volumen, en ml, de solución patrón de fluoruro añadida.
C = concentración, en mg/l, de la solución patrón adicionada.
S = pendiente del electrodo.
ΔE = diferencia de los potenciales E1 y E2 obtenidos en 39(b).4.2.
Este valor se deberá multiplicar por el factor de dilución correspondiente, en caso de haber sido necesario efectuarla.
39(b).6 Observaciones.
39(b).6.1 Todas las soluciones deben conservar una temperatura próxima a 25 °C ± 1 °C durante la medida.
39(b).7 Referencias.
1. H. E. Haller y C. H. Junge, 1974, O. I. V. 317/FV/518 y 133/FV/439.
2. Deschreider, A., y Mme. R. Meaux, 1974. Determination des flúor dans les vins et eaux minerales naturelles au moyen d’une electrode ionique specifique.
48. RELACIÓN P/α
48.1 Principio.
Estimación de los contenidos relativos de glucosa y fructosa, en mostos, vinos dulces y mistelas, mediante el establecimiento de la relación entre azúcares reductores y desviación polarimétrica a 20 °C.
48.2 Material y aparatos.
48.2.1 Como en 7(a).2; 7(b).2 o 7(c).2.
48.2.2 Polarímetro con luz monocromática amarilla (lámpara vapor de sodio) y tubo de 20 cm.
48.3 Procedimiento.
48.3.1 Determinar el contenido en azúcares reductores según el método 7(a), 7(b) o 7(c).
48.3.2 Medida de la desviación polarimétrica.
Una vez verificado el correcto reglaje del polarímetro, comprobando la correspondencia del valor «cero» con la existencia de un campo luminoso uniforme, ajustándolo si es necesario, introducir en el tubo del polarímetro el líquido de defecación sin diluir, incoloro y limpio, utilizando para la determinación de azúcares reductores, evitando la formación de burbujas de aire. Anotar la temperatura del líquido que debe ser lo más próxima posible a 20°C.
Efectuar la lectura de la desviación polarimétrica varias veces sucesivas, tomando la media de los resultados obtenidos.
48.4 Cálculos.
Determinar el poder rotatorio y la relación P/oc aplicando las fórmulas siguientes:
OC20 = oct + oct (t ‒ 20) x K
P/OC20 (expresado con su correspondiente signo)
Siendo:
P = g/l de azúcares reductores.
oct = poder rotatorio en divisiones sacarimétricas a t °C (un grado polarimétrico = 4,615 divisiones sacarimétricas).
OC20 = poder rotatorio en divisiones sacarimétricas a 20 °C.
t = temperatura del líquido en el momento de la lectura.
K = coeficiente variable con la naturaleza de los azúcares, dado por la tabla I en función del valor P/oct.
48.5 Referencias.
1. J. Blouin. Manual practique d’analyse des Moúts et Vins. 1977.
2. J. Ribereau Gayón, E. Peynaud. Sciences et Techniques du Vin. Tome I. 1972.
TABLA I
| P/oct | K |
|---|---|
| ‒ 1,19 | 0,006 |
| ‒ 1,41 | 0,007 |
| ‒ 1,73 | 0,008 |
| ‒ 2,24 | 0,009 |
| ‒ 2,64 | 0,012 |
| ‒ 3,20 | 0,012 |
| ‒ 4,06 | 0,013 |
| ‒ 5,56 | 0,014 |
| ‒ 8,8 | 0,022 |
| ‒ 21 | 0,042 |
VINAGRES
9. METANOL, METODO DEL ÁCIDO CEOMOTRÓPICO
9.1 Principio.
Como en 37.1 de los métodos oficiales de análisis de vinos.
9.2 Material y aparatos.
Como en 37.2 de los métodos oficiales de análisis de vinos.
9.3 Reactivos.
9.3.1 Como 37.3.1 de los métodos oficiales de análisis de vinos.
9.3.2 Ídem.
9.3.3 Ídem.
9.3.4 Etanol de 96° (v/v).
9.3.5 Matraz aforado de 25 ml.
9.4 Procedimiento.
Tomar 25 ml de vinagre, destilar en destilador simple y recoger 15 ml de destilado en matraz aforado de 25 ml sumergido en baño de hielo. Añadir el etanol suficiente para que una vez enrasado a 25 ml con agua destilada la concentración del etanol sea del 5-6 por 100 (9.6.1). Continuar como en 37.4 de los métodos oficiales de análisis de vinos.
9.5 Cálculo.
Como en 37.5 de los métodos oficiales de análisis de vinos.
9.6 Observaciones.
9.6.1 En vinagres normales suele ser suficiente añadir 1,5 por 100 de alcohol de 96 por 100.
9.7 Referencias.
1. Association of Official Agricultural Chemists Official Methodos of Analysis, 1965, página 138.
10(a). ÁCIDO TARTÁRICO
(Método cualitativo)
10(a).1 Principio.
Precipitación del ácido tartárico en forma de bitartrato potásico.
10(a).2 Material y aparatos.
10(a).2.1 Centrífuga.
10(a).3 Reactivos.
10(a).3.1 Ácido acético concentrado puro.
10(a).3.2 Solución de precipitación.
Disolver 300 g de cloruro de potasio puro, 5,75 g de tartrato de sodio y potasio (sal de Seignette) y 1,25 g de oxalato de potasio y enrasar a 1.000 ml con agua destilada.
A baja temperatura la solución deposita pequeños cristales, por lo que se debe de calentar ligeramente antes de emplearse.
10(a).3.3 Etanol de 96°.
10(a).4 Procedimiento.
Poner en un tubo de centrífuga 10 ml de vinagre, 0,4 ml de ácido acético 7,5 ml de la solución de precipitación y 2 ml de etanol. Agitar con una varilla de vidrio hasta que aparezca un enturbiamiento cristalino aproximadamente a los diez segundos. Mantener el tubo durante una hora a 8 ºC aproximadamente. Al cabo de este tiempo agitar enérgicamente el contenido del tubo con una varilla de vidrio, con la precaución de mezclar bien con la solución que sobrenada. Dejar en reposo aproximadamente a 8 ºC durante una hora. Transcurrido este tiempo centrifugar.
10(a).5 Interpretación de resultados.
La aparición de precipitado indica la presencia de ácido tartárico en la muestra.
10(a).6 Referencias.
1. Recueil des Méthodes Internationales d’Analyse des Vins. O. I. V. A-12-h.
10(b). ÁCIDO TARTÁRICO
(Método cuantitativo)
10(b).1 Principio.
Aislamiento del ácido tartárico mediante columna de resinas cambiadoras de aniones y posterior valoración clorimétrica.
10(b).2 Material y aparatos.
10(b).2.1 Resinas aniónicas de basicidad fuerte (III Merck o equivalente). Las resinas deben de encontrarse en forma de acetato. Para ello deben estar al menos veinticuatro horas en contacto con una solución de ácido acético al 30 por 100.
10(b).2.2 Columnas de vidrio de 10-11 mm de diámetro y 30 cm de longitud con llave en la parte inferior.
10(b).3 Reactivos.
10(b).3.1. Ácido acético al 30 por 100 (v/v).‒Introducir 300 mililitros de ácido acético glacial en un matraz aforado y completar a 1.000 ml con H2O destilada.
10(b).3.2 Ácido acético al 0,5 por 100 (v/v).‒Introducir 5 mililitros de ácido acético glacail en un matraz aforado y completar hasta 1.000 ml con H2O destilada.
10(b).3.3 Solución de sulfato de sodio al 7,1 por 100 (0,5 N).‒Disolver 71 g de sulfato de sodio anhidro (Na2SO4) y completar con agua destilada hasta 1.000 ml.
10(b).3.4 Reactivo vanádico.‒Disolver 10 g de metavanadato de amonio en 150 ml de una solución de NaOH N. Pasar esta solución a un matraz aforado de 500 ml, añadir seguidamente 200 ml de una solución de acetato de sodio al 27 por 100 y llevar luego a 500 ml con H2O destilada.
10(b).3.5 Solución de SO4H2 2 N.
10(b).3.6 Solución de SO4H2 0,1 N.
10(b).3.7 Solución de ácido peryódico 0,1 N (0,05 M).‒Introducir 10,696 g de peryodato de sodio en un matraz aforado de 1.000 ml, añadir 50 ml de SO4H2 N y completar hasta 1.000 mililitros con H2O destilada.
10(b).3.8 Solución de glicerol al 10 por 100.‒Pesar 10 g de glicerol muy puro bidestilado y llevarlo a 100 ml con H2O destilada.
10(b).3.9 Solución de ácido tartárico de 1 g por litro.‒En un matraz aforado de 500 ml disolver 500 mg de ácido tartárico (puro) en 6,6 ml de sosa N y enrasar con solución de sulfato de sodio al 7,1 por 100 a 500 ml.
10(b).4 Procedimiento.
10(b).4.1 Preparación de las columnas de resinas aniónicas. Colocar en el fondo de la columna [10(b).2.2] lana de vidrio formando tapón de 2-3 mm de altura, aproximadamente, y añadir agua hasta una altura de unos 5 mm sobre el tapón de lana de vidrio. Añadir después al resina cambiadora de aniones, conservada en ácido acético al 30 por 100, agitar esta suspensión y agregar rápidamente un volumen de unos 10 ml, evitando la formación de burbujas de aire, depositando en la superficie un tapón de lana de vidrio, que se conduce con una varilla de vidrio, con el fin de no remover las resinas en los sucesivos lavados anteriores en su superficie. Las resinas solamente pueden servir para una sola vez.
10(b).4.2 Aislamiento del ácido tartárico.‒Abrir la llave inferior de la columna y dar salida al ácido acético diluido hasta unos 2-3 ml por encima del tapón de lana superior. Añadir seguidamente unos.10 ml de ácido acético al 0,5 por 100 y vaciar hasta igual altura; efectuar estos lavados cuatro veces.
Después del último lavado y con la llave cerrada, adicionar 10 ml de vinagre a analizar y señalar la altura alcanzada con un lápiz graso, abrir a continuación la llave y dejar salir vinagre goteando a razón de 1-1,5 gotas por segundo (25-30 ml por minuto) hasta un poco por encima del tapón superior de lana vidrio. Llenar de nuevo la columna con ácido acético al 0,5 por 100 hasta la señal del lápiz graso y dejar salir a la misma velocidad que la vez anterior y lavar después en la misma forma siete veces con 10 ml de agua destilada.
Al terminar el último lavado, cerrar la llave cuando el nivel del líquido se encuentre un poco más arriba del tapón de lana de vidrio superior.
Colocar un matraz receptor aforado de 100 ml. Eluir los ácidos fijados en el cambiador de aniones, mediante adiciones de solución de sulfato de sodio 110(b).3.31 hasta la señal trazada en el tubo.
Para hacer esta operación resulta práctico colocar un frasco con solución de sulfato unido por el cuello a la columna mediante un manguito de caucho y con unas pinzas para regular la, caída del líquido en la bureta o tubo. Puesto así en comunicación los dos aparatos (frasco y bureta o tubo), abrir las pinzas del caucho de unión y la llave inferior, dejando caer el líquido a la bureta hasta unos 10 cm de altura y sin que queden huecos vacíos. Regular la salida del líquido en la proporción que queden huecos vacíos. Regular la salida del líquido en la proporción de 2-3 gotas por segundo, para llenar el matraz receptor hasta el enrase.
10(b).4.3 Determinación del ácido tartárico.‒En dos matraces cónicos de 100 ml (A) y (B) introducir 20 ml del eluido. El matraz (A) sirve para la medida y el matraz (B), en el cual el ácido tartárico es destruido por el ácido pervódico, constituye el testigo.
Añadir al matraz (A) 2 ml de SO4H2 2 N, 5 ml de SO4H2 0,1 N y 1 ml de glicerol al 10 por 100.
En el matraz (B), añadir 2 ml de SO4H2 2 N, 5 ml de ácido peryódico 0,1 N, esperar quince minutos y añadir 1 ml de solución de glicerol al 10 por 100 para destruir el exceso de ácido peryódico. Esperar dos minutos.
Verter a continuación, agitando, primero en el matraz (B) e inmediatamente después en el matraz (A). 5 ml del reactivo vanádico. Disparar inmediatamente un cronómetro e introducir el contenido de dichos matraces en las cubetas de caras paralelas de lo mm de espesor del espectrofotómetro. Al cabo de un minuto y treinta segundos medir la densidad óptica a 490 nm del líquido procedente del matraz (A) (medida) después de haber reglado el aparato para obtener la transmisión al 100 por 100 con la cubeta conteniendo el líquido testigo (B).
10(b).5 Cálculo.
10(b).5.1 Preparación de la curva patrón.‒Tomar 10, 20, 30, 40 y 50 ml de la solución 10(b).3.9 e introducir cada alícuota en un matraz aforado de 100 ml. Enrasar con solución de sulfato de sodio al 7,1 por 100. Las diferentes diluciones corresponden a eluidos del vinagre conteniendo 1, 2, 3, 4 y 5 g de ácido tartárico por litro.
En dos matraces (A) y (B) de 100 ml poner 20 ml de cada una de estas soluciones y tratarlas idénticamente igual a los eluidos del vinagre anteriormente descrito.
La representación gráfica de las densidades ópticas de estas soluciones está en función de la cantidad de ácido tartárico. Para ello es recomendable y más preciso partir de concentraciones 0,1; 0,2; 0,3; 0,4: 0,5; 0,6; 0,7; 0,8; 0,9 y 1 g/l de ácido tartárico.
10(b).5.2 Comprobar los valores obtenidos en el ensayo con los correspondientes de la curva patrón.
10(b).6 Referencias.
1. Recueil des Méthodes d’Analyse des Vins Al2h.
11. COBRE
11.1 Principio.
Determinación directa por espectrofotometría de absorción atómica.
11.2 Material y aparatos.
Espectrofotómetro de absorción atómica, con lámpara específica de cobre.
11.3 Reactivos.
11.3.1 Solución patrón de 1.000 mg/l de Cu.‒Disolver 1.000 g de cobre puro en el mínimo volumen necesario de NO3H (1 + 1) y diluir a 1 litro con NO3H al 1 por 100 (v/v).
11.3.2 Soluciones que contengan de 1 a 10 mg/l de Cu.‒Diluir partes alícuotas de la solución patrón con ácido acético al 5 por 100
11.4 Procedimiento.
11.4.1 Calibrado del aparato.‒Ajustar el aparato a una longitud de onda de 324,7 nm en condiciones tales que se obtenga una respuesta lineal al introducirse las series de patrones preparados.
11.4.2 Determinación.‒Efectuar la lectura directa de la absorbancia.
11.5 Interpretación de resultados.
Partiendo de los valores de absorbancia obtenidos, hallar mediante la curva patrón las concentraciones de cobre de la muestra.
11.6 Referencias.
1. G. Charlot et D. Bezier: «Analyse quantitative mlnerale». El Masson. París (1955). 749-752.
2. H. E. Parker: «Atomic Absorption Newlette» (1963), 13.
3. Standart conditionc for Zn and Cu. Analytical Methods for atomic absorption spectrophotometry «Perkin-Elmer».
4. F. Rouselet: «Spectrophotometrie par absorption atomique Boudin». Ed. París (1968), págs. 59-144.
12. CLORUROS
12.1 Principio.
Determinación directa de cloruros por potenciometría utilizando el electrodo Ag/AgCl.
12.2 Material y aparatos.
12.2.1 pH-metro con escala que permita apreciar 2 mV.
12.2.2 Agitador magnético.
12.2.3 Electrodo Ag/AgCl con una solución saturada de nitrato de potasio como electrolitro.
12.2.4 Microbureta graduada en 1/100 de mililitro.
12.3 Reactivos.
12.3.1 Solución patrón de cloruros.‒Disolver 2,1027 g de cloruro de potasio puro para análisis (máximo 0,005 por 100 de bromo), previamente desecado, en agua destilada hasta 1 litro, 1 ml de esta solución contiene 1 mg de ion cloro.
12.3.2 Solución de nitrato de plata.‒Disolver 4,7912 g de nitrato de plata en una solución alcohólica al 10 por 100 (v/v) hasta 1 litro; 1 ml de esta solución corresponde a 1 mg de ion cloro.
12.3.3 Ácido nítrico concentrado (d = 1,40).
12.4 Procedimiento
12.4.1 Determinación del potencial del punto de equivalencia.‒Introducir 5,0 ml de la solución patrón de cloruros en un vaso de 150 ml. Diluir aproximadamente a 100 ml con agua destilada y acidificar con 1.0 ml de ácido nítrico (12.3.3). Introducir los electrodos. Añadir la solución de nitrato de plata agitando moderadamente
Los cuatro primeros mililitros se adicionan en fracciones de 1 ml y se leen las correspondientes lecturas en milivoltios; los dos siguientes mililitros, en fracciones de 0,2 ml y se continúa adicionando en fracciones de 1 ml hasta que se hayan alcanzado un total de 10 ml. Después de cada adición se debe esperar unos treinta segundos antes de hacer la correspondiente lectura en milivoltios. Los valores así obtenidos se llevan sobre un papel milimetrado en función de los correspondientes mililitros de solución de nitrato de plata, y a partir del punto singular de la curva obtenida, determinar el potencial del punto de equivalencia.
Para comprobar el potencial del punto de equivalencia, llevar a un vaso de 150 ml, 5,0 ml de la solución patrón de cloruros, 95 ml de H2O destilada y 1 ml de ácido nítrico concentrado. Introducir el electrodo y valorar agitando, justamente hasta el potencial del punto de equivalencia.
Repetir esta operación hasta obtener una buena concordancia de resultados
Este control debe de efectuarse antes de determinar la concentración de cloruros en cada serie de muestras a analizar.
12.4.2 Determinación.‒Llevar a un vaso de 150 ml 50,0 ml de vinagre. Añadir 50 ml de H2O destilada y 1 ml de ácido nítrico concentrado. Valorar a continuación, siguiendo el procedimiento descrito anteriormente, hasta alcanzar el potencial del punto de equivalencia.
12.5 Cálculos.
La cantidad de cloruros se determina mediante las expresiones:
0,02 n en gramos de ion cloro.
0,5633 n en miliequivalentes por litro de ion cloro.
0,0329 n en gramos de cloruro sódico por litro.
Siendo:
n = volumen, en ml de solución de nitrato de plata necesario para alcanzar el potencial del punto de equivalencia.
12.6 Referencias.
1. Recueil des Méthodes Internationales d’Analyse de Vins. OIV A-15j.
13. GRADO ALCOHÓLICO
13.1 Principio.
Separación del alcohol por destilación, oxidación del mismo por dicromato potásico y determinación del contenido por valoración del exceso de dicromato.
13.2 Material y aparatos.
Como en los apartados 5(a).2.1; 5(a).2.1.1; 5(a).2.1.2; 5(a).2.1.3 y 5(a).2.1.4 de los métodos Oficiales de Análisis de Vinos.
13.3 Reactivos.
13.3.1 Solución de dicromato potásico.‒Disolver 33,608 g de dicromato potásico puro en una cantidad suficiente de agua destilada y completar hasta 1 litro a 20 °C. Esta solución corresponde al grado alcohólico internacional O. I. V.
1 ml de esta solución óxida 7,8934 mk de alcohol.
13.3.2 Solución de sulfato de hierro y de amonio. Disolver 135 g de sulfato ferroso amoniacal y 20 ml de ácido sulfúrico puro en una cantidad de agua suficiente y completar hasta 1 litro, un volumen de esta solución corresponde a medio volumen de solución de dicromato potásico cuando está recientemente preparada. Esta solución se oxida lentamente, por lo que debe ser valorado frecuentemente en la misma que se realiza la determinación del alcohol, sustituyendo la disolución alcohólica por agua destilada.
13.3.3 Solución de permanganato potásico.‒Disolver 1,088 gramos de permanganato potásico en una cantidad suficiente de agua y completar hasta 1 litro.
13.3.4 Solución de ácido sulfúrico diluido.‒A 500 ml de agua destilada añadir, poco a poco y agitando, 500 ml de ácido sulfúrico puro. Después de enfriado, completar hasta 1 litro con agua destilada.
13.3.5 Reactivo de ortofenantrolina ferrosa.‒Disolver 0,695 gramos de SO4 Fe 7H2O en 100 ml de agua, añadir 1,485 gramos de monohidrato de ortofenantrolina. Calentar para favorecer la disolución.
13.3.6 Lechada de cal 4N.
13.4 Procedimiento.
13.4.1 Destilación.‒Medir 200 ml de vinagre en matraz aforado y anotar la temperatura. Introducir el vinagre en el matraz de destilación que contenga unos fragmentos de material poroso. Lavar el matraz cuatro veces con 5 ml de agua. Alcalinizar con un ligero exceso de lechada de cal 4N. Recoger el destilado en matraz aforado de 100 ml. Enrasar con agua a la misma temperatura que se midió el vinagre inicialmente.
13.4.2 En un matraz Erlenmeyer, con tapón esmerilado, de 250 ml, poner 20 ml de solución valorada de dicromato potásico, 20 ml de ácido sulfúrico diluido y agitar. Añadir 10 ml de destilado exactamente medidos. Tapar el Erlenmeyer, agitar y esperar por lo menos treinta minutos, agitando de vez en cuando.
13.4.3 Valoración.‒Valorar el exceso de dicromato con la solución de sulfato ferroso amónico. Cuando la coloración verde de la solución vire a verde azulado, añadir cuatro gotas de reactivo de ortofenantrolina. Detener la adición de solución ferrosa cuando el líquido vire a marrón.
A menudo se puede llegar a pasar el viraje, siendo entonces necesario volver al punto preciso, adicionando solución de permanganato potásico. Un décimo del volumen empleado de esta solución se resta del volumen empleado de sulfato ferroso.
Hacer una prueba en blanco sustituyendo los 10 ml de destilado por agua.
13.5 Cálculos.

Siendo:
n = volumen, en ml, gastados en la valoración.
n’ = volumen, en ml, gastados en la prueba en blanco.
13.6 Observaciones.
Si el grado alcohólico del vinagre es superior a 0,8°, repetir la oxidación utilizando 10 ml de destilado diluido a 1/2.
13.7 Referencias.
1. Recueil des Méthodes Internationales d’Analyse des Vins. O. I. V. A-2-16.
14. ARSÉNICO
14.1 Principio.
Reducción del arsénico a arsenamina gaseosa que reacciona con dietilditiocarbamato de plata (disuelto en piridina), dando un complejo de color rojo.
14.2 Material y aparatos.
14.2.1 Aparato Quickfit (figura I).
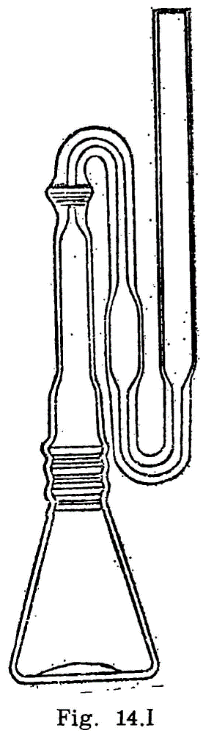
14.2.2 Espectrofotómetro o colorímetro que permita lecturas a 538 nm.
14.3 Reactivos.
14.3.1 Solución de dietilditiocarbamato de plata.‒Disolver 1.000 g de este reactivo en 200 ml de piridina para cromatografía. Esta solución puede conservarse algunas semanas.
14.3.2 Acetato de plomo en algodón.‒Impregnar el algodón hidrófilo en solución acuosa de acetato de plomo al 10 por 100, escurrir el líquido en exceso y desecar al vacío.
14.3.3 Solución acuosa de ioduro potásico al 15 por 100.
14.3.4 Solución acuosa de sulfato de cobre al 2 por 100.
14.3.5 Solución de cloruro estannoso.‒Disolver 0,33 g de cloruro estannoso en 100 cc de ClH al 32 por 100 (d = 1,16).
14.3.6 Zn puro granulado, exento de arsénico.
14.3.7 Ácido nítrico (d = 1,40).
14.3.8 Ácido sulfúrico (d = 1,84).
14.3.9 Solución de 100 mg/l de arsénico.
14.4 Procedimiento.
14.4.1 Preparación de la muestra.‒En un matraz Kjeldahl de 500 ml introducir 100 ml de vinagre con 100 ml de NO3H (d = 1,40), para evitar las pérdidas de As. Llevar a ebullición hasta reducir el volumen a 10 ml, añadir 5 ml de ácido sulfúrico (d = 1,84) y continuar la ebullición adicionando NO3H gota a gota hasta la total decoloración. Después de la expulsión de los vapores nitrosos dejar enfriar, diluir con 10 ml de agua destilada y calentar hasta casi sequedad. Una vez frío el matraz, trasvasar el residuo al matraz de valoración utilizando 40 ml de agua destilada.
14.4.2 Valoración.‒Poner en el tubo en U, 3 ml de la solución de dietilditiocarbamato de plata. Colocar en la parte superior del tubo intermedio del aparato el algodón impregnado en acetato de plomo con objeto de retener el sulfídrico y la fosfamina que puedan desprenderse al mismo tiempo que la arsenamina.
Introducir la muestra preparada en el Erlenmeyer de 100 ml del aparato descrito y añadir sucesivamente 10 ml de la solución de cloruro estannoso, 5 ml de la solución de ioduro potásico y 1 ml de la solución de sulfato de cobre. Después de quince minutos añadir 5 gramos de Zn granulado y cerrar rápidamente el aparato. Colocarlo en la oscuridad y dejar pasar vapores por lo menos durante una hora.
14.4.3 Preparación de la curva patrón.‒A partir de una solución 14.3.9., preparar soluciones que contengan 0,5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 μg de arsénico por litro.
14.5 Cálculos.
Calcular el contenido en arsénico utilizando la correspondiente curva patrón.
14.6 Observaciones.
14.6.1 Interfiere en la determinación el antimonio cuyo complejo absorbe a 510 nm.
14.6.2 La Ley de Lambert-Beer se cumple entre las concentraciones de 0 y 15 microgramos de As.
14.6.3 Este método tiene un límite de detección de 0,2 microgramos.
14.7 Referencias.
1. Vasak. V y Sedivec V (1952) Chem. Listy 46, 341, citado por M. D. Garrido, M. L Gil y C. Llaguno. An. Bromatología XXVI-2 (1974), 167-176.
2. Recueil des Méthodes Internationales d’Analys des Vins. O. I. V. A-34 f.
15. CINC
15.1 Principio.
Determinación directa por espectrofotometría de absorción atómica.
15.2 Material y aparatos.
15.2.1 Espectrofotómetro de absorción atómica.
15.2.2 Lámpara de cinc.
15.3 Reactivos.
15.3.1 Solución patrón de 1.000 ppm de Zn.‒Disolver 1.000 gramos de Zn puro en el mínimo volumen necesario de ácido nítrico (1 + 1) y diluir a 1 litro con ácido nítrico al 1 por 100 (v/v).
15.4 Procedimiento.
15.4.1 Preparación de la muestra.‒No se necesita preparación especial. En caso de elevada concentración, realizar una o varias diluciones hasta quedar dentro del rango de medida.
15.4.2 Determinación.‒Diluir partes alícuotas de la solución (15.3.1) con ácido acético al 5 por 100 para obtener soluciones de 0, 0,5, 1, 1,5 y 2 ppm. Aspirar las soluciones patrón y a continuación la muestra, anotando las absorbancias obtenidas a 213,8 nm.
15.5 Interpretación de resultados.
Partiendo de los valores de absorbancia obtenidos, hallar las concentraciones de cinc de la muestra.
15.6 Referencias.
1. H. E. Parker, Atomic Absorption Newsletter (1963), 13.
2. Standar conditions for Zn and Cu. Analytical Methods for atomic absorption spectrophotometry «Perkin-Elmer».
6. ACETILMETILCARBINOL (ACETOÍNA)
6.1 Principio.
Determinación cuantitativa de la acetoína por cromatografía de gases.
6.2 Material y aparatos.
6.2.1 Equipo de cromatografía de gases compuesto por:
6.2.1.1 Detector de ionización de llama.
6.2.1.2 Horno con temperatura regulable.
6.2.1.3 Registrador.
6.2.1.4 Gases: Aire-nitrógeno.
6.2.1.5 Columna: FFAP 2,5 por 100 sobre Chromosorb G (HP), añadiéndose un 0,5 de un reductor de colas (p. e., Carbowax 1.500) y de dimensiones 2 metros de longitud y 1/8 de pulgada de diámetro externo de la columna.
6.2.2 Microjeringa de 1 a 10 μl.
6.3 Reactivos.
6.3.1 Acetoína.
6.3.2 Pentanol-1.
6.3.3 Alcohol etílico absoluto.
6.3.4 Hidróxido cálcico.
6.4 Procedimiento.
6.4.1 Disoluciones patrón.
6.4.1.1 De acetoína: A partir de acetoína purificada de su posible contenido en diacetilo mediante destilación, obtener soluciones de acetoína que cubran un rango de concentraciones de 10 a 500 mg/l en agua destilada.
6.4.1.2 De patrón interno: Añadir 2 ml de pentanol-1 en 100 ml de solución hidroalcohólica al 50 por 100.
6.4.2 Determinación.
6.4.2.1 Condiciones cromatográficas.
Gas portador: Nitrógeno (flujo de 12,5 mg/min).
Temperatura horno: 70 °C.
Temperatura inyector: 180 °C.
Inyección: 4 o 2 μl.
Patrón interno: Pentanol-1.
6.4.2.2 Método operatorio.
A cada una de las soluciones patrón de acetoína preparadas en 6.4.1.1 añadir solución de pentanol-1, de tal forma que las soluciones que contengan de 10 a 50 mg/l de acetoína se le añadan 15 μl y a las que contengan de 50 a 500 mg/l se le añadan 35 μl, e inyectarlas a continuación, obteniendo la curva de calibrado (se deben efectuar dos curvas de calibrado, una que cubra el rango de 0-50 mg/l de acetoína y otra de 50 a 300 mg/l).
La inyección de la muestra puede efectuarse directamente del vinagre una vez añadido el patrón interno, pero teniendo en cuenta que el tiempo de retención es elevado y la concentración del acético es muy grande, obliga a esperar un largo período de tiempo entre dos inyecciones sucesivas. Para evitar esta dificultad, deben neutralizarse a pH = 7 las muestras, preferiblemente con Ca(OH)2 sólido, para evitar variaciones de volumen. Téngase en cuenta que la concentración de acetoína en el medio neutro puede variar con el tiempo, por lo que la neutralización debe hacerse inmediatamente antes de inyectar en el cromatógrafo.
6.5 Cálculos.
Los cromatogramas obtenidos de las distintas soluciones patrón permiten representar gráficamente la relación existente entre el cociente:

La relación de áreas obtenida para la muestra se compara con la curva de calibrado (concentración de acetoína/relación de áreas), obteniéndose la cantidad de acetoína de la muestra.
6.6 Referencias.
1. E. F. Gorostiza, M. L. Gil de la Peña y M. C. Gómez Cordobés. Instituto de Fermentaciones Industriales.
16. PROLINA
16.1 Principio.
Determinación cuantitativa de la prolina por reacción con ninhidrina en medio ácido.
16.2 Material y aparatos.
16.2.1 Tubos de ensayo con tapón de rosca.
16.2.2 Espectrofotómetro o colorímetro que permita efectuar lecturas a 517 nm.
16.3 Reactivos.
16.3.1 Ninhidrina al 3 por 100 en éter monometílico del etilenglicol.
16.3.2 Ácido fórmico.
16.3.3 Isopropanol agua 1:1 (v/v).
16.4 Procedimiento.
Diluir la muestra hasta que contenga de 0,05 a 0,50 milimoles/ml de prolina. Tomar 1/2 ml de muestra diluida a introducirla en un tubo de ensayo con tapón de rosca, añadir 0,25 ml de ácido fórmico y 1 ml de solución de ninhidrina al 3 por 100. Cerrar herméticamente el tubo e introducirlo en un baño de agua hirviendo durante catorce o quince minutos. Enfriarlo a unos 20 °C de cinco a diez minutos, añadiendo mientras se enfría 5 ml de solución de isopropanol-agua 1:1 (v/v).
A continuación efectuar la lectura de la absorción a 517 nm después de cinco minutos y antes de treinta minutos.
Efectuar un blanco siguiendo el procedimiento anterior, pero usando 0,5 ml de agua destilada en vez de la solución problema.
16.5 Interpretación de resultados.
Calcular el contenido en prolina por comparación con una curva patrón que comprenda concentraciones entre 0,05 y 0,50 milimoles/ml.
16.6 Referencias.
1. C. S. Ough J. of Food Science 34(3), pág. 228 (1969).
ORUJOS, HECES Y LÍAS
1. CONTENIDO ALCOHÓLICO EN ORUJOS, HECES Y LÍAS
1.1 Principio.
Se determina por destilación de la muestra alcalinizada y medida del contenido alcohólico por alcohometría.
1.2 Material y aparatos.
1.2.1 Aparato de destilación.‒Consta de las siguientes partes:
1.2.1.1 Matraz de destilación de 1.000 ml con rodaje esmerilado. Se puede utilizar de boca ancha o de boca estrecha.
1.2.1.2 Columna de rectificación de 20 cm de largo.
1.2.1.3 Disco metálico o de amianto con un orificio de 8 cm de diámetro.
1.2.1.4 Refrigerante de West de 40 cm de longitud con circulación rápida de agua.
1.2.2 Equipo de medida.
1.2.2.1 Alcohómetros.
1.2.2.2 Termómetros.‒Graduados en grados y décimas de grado y comprendiendo el intervalo de 0-30°.
1.2.2.3 Probeta cilíndrica de 36 mm de diámetro y 320 mm de altura.
1.2.3 Granatorio.
1.2.4 Matraz aforado de 200 ml.
1.2.5 Vaso de precipitados.
1.3 Reactivos.
1.3.1 Lechada de cal-solución de 120 g de CaO en 1.000 ml de agua.
1.3.2 Ácido sulfúrico al 10 por 100.
1.4 Procedimiento.
El producto a analizar es vertido en un vaso de precipitados donde es agitado para su homogeneización y al mismo tiempo para favorecer la expulsión de CO2 que pueda contener.
En el caso de orujos, el muestreo se realiza de forma que la muestra sea lo más homogénea posible.
Pesar 200 g de muestra homogeneizada y vaciar en el matraz de destilación añadiendo 200 ml de agua, parte de los cuales se utilizan para enjuagar el vaso que ha contenido la muestra, y a continuación se añade lechada de cal hasta neutralizar en exceso (10 ml aproximadamente).
Destilar a continuación recogiendo 200 ml, o bien las tres cuartas partes de este volumen (150 ml).
Añadir al destilado más gotas de una disolución acuosa de tornasol y comprobamos que el destilado toma color azul, de franca alcalinidad, debida al NH3 que contiene.
Seguidamente, con una pipeta se adiciona ácido sulfúrico al 10 por 100 en cantidad suficiente para que el destilado vire a rojo, añadiendo un poco más (0,5 ml) para que el medio quede francamente ácido.
Completar con agua destilada el contenido de los 200 ml del matraz y se procede a una segunda destilación, como si de un vino se tratase.
El destilado recogido de esta segunda destilación está ya en condiciones para determinar su grado alcohólico por aerometría y efectuar la corrección a 20 °C.
1.5 Expresión de los resultados.
El resultado se puede expresar:
‒ cc de alcohol absoluto/100 g de muestra
1.6 Referencias.
1. Recueil des Méthodes Internationales d’Analyse des vins O.I.V. 1969, A2 1-19.
2. De Bernardi, P.; García Viana, E. 1969. «Determinación de grado alcohólico de heces y lías». Estación Enológica de Requena.
Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid




