Agencia Estatal Boletín Oficial del Estado






El trasplante de células y tejidos humanos es un área de la Medicina que ha experimentado un enorme crecimiento en los últimos años y está proporcionando grandes posibilidades terapéuticas para muchos pacientes.
Su creciente utilización clínica requiere la aprobación de una norma que participe de los principios de voluntariedad, anonimato entre donante y receptor, altruismo y solidaridad que caracterizan el modelo de trasplantes del Sistema Nacional de Salud y que recoja los avances técnicos y científicos producidos en esta materia, al tiempo que prevea los sistemas de control de los procesos que se suceden desde la obtención de las células y tejidos hasta su implantación, y las condiciones que deben reunir los centros y unidades de obtención y aplicación y los establecimientos de tejidos. Todo ello con el objetivo de asegurar la calidad y la seguridad de las células y tejidos utilizados que eviten la transmisión de enfermedades y faciliten su utilización terapéutica.
Además, este real decreto contempla que la disponibilidad de células y tejidos humanos con fines terapéuticos depende, en gran medida, de la disposición de los ciudadanos a hacer efectivas las donaciones, de ahí que, como se ha recomendado reiteradamente a los Estados miembros desde las instituciones de la Unión Europea, se promueva la existencia de sistemas y canales de información precisos sobre la donación de estas células y tejidos, así como de criterios transparentes y objetivos de acceso a estas células y tejidos sobre la base de una evaluación objetiva de las necesidades médicas, y se fomente una participación destacada del sector público y de las organizaciones sin ánimo de lucro en la prestación de los servicios de utilización de células y tejidos humanos.
En el ámbito de la Unión Europea estos objetivos, así como la necesidad de asegurar regulaciones nacionales armonizadas en esta materia, han quedado plasmados en la Directiva 2004/23/CE del Parlamento Europeo y del Consejo, de 31 de marzo de 2004, relativa al establecimiento de normas de calidad y de seguridad para la donación, la obtención, la evaluación, el procesamiento, la preservación, el almacenamiento y la distribución de células y tejidos humanos, y en la Directiva 2006/17/CE de la Comisión, de 8 de febrero de 2006, por la que se aplica la Directiva 2004/23/CE del Parlamento Europeo y del Consejo en lo relativo a determinados requisitos técnicos para la donación, la obtención y la evaluación de células y tejidos humanos. Este real decreto incorpora a nuestro ordenamiento jurídico sus contenidos.
Los principios de este real decreto se deben aplicar a todos los tejidos y células humanas, incluyendo las células progenitoras hematopoyéticas de sangre periférica, cordón umbilical o médula ósea; las células reproductoras, excepto en los aspectos regulados en la Ley 14/2006, de 26 de mayo, sobre técnicas de reproducción humana asistida; las células y tejidos fetales, y las células troncales adultas y embrionarias cuando su finalidad sea el uso terapéutico o la aplicación clínica.
Quedan excluidos, sin embargo, la sangre y los productos sanguíneos, a excepción de las células progenitoras hematopoyéticas y los órganos humanos. Tampoco cubre los procedimientos de investigación con células y tejidos que no incluyan una aplicación en el cuerpo humano (investigación in vitro o en modelos animales), ya que sólo se exigirán las normas de calidad y de seguridad que en el real decreto se recogen, a aquellos tejidos y células que se utilicen en ensayos clínicos con aplicaciones en seres humanos.
Esta norma prevé, además, la posibilidad de que existan establecimientos entre cuyas actividades figure la preservación de células y/o tejidos para un eventual uso autólogo. Aunque no existe una base científica actual ni respaldo de las instituciones europeas a dicha práctica, se ha considerado necesario regularla dada la presencia y progresiva implantación de este tipo de establecimientos en los países de nuestro entorno. Este real decreto establece las condiciones que tales establecimientos deben cumplir.
En la redacción de este real decreto se ha tenido en cuenta la Carta de Derechos Fundamentales de la Unión Europea y el Convenio del Consejo de Europa para la protección de los derechos humanos y la dignidad del ser humano respecto de las aplicaciones de la biología y la medicina, suscrito en Oviedo el día 4 de abril de 1997, y que entró en vigor en España el 1 de enero de 2000. Desde el punto de visto del ordenamiento jurídico interno, en el tratamiento de los datos personales que sean procesados en aplicación de los principios desarrollados en este real decreto se ha considerado lo dispuesto en la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal, y desde el punto de vista del régimen de los derechos que pudieran verse afectados, la referencia necesaria ha sido la Ley 41/2002, de 14 de noviembre, básica reguladora de la autonomía del paciente y de derechos y obligaciones en materia de información y documentación clínica.
En la elaboración de este real decreto se ha tenido en cuenta la Ley 30/1979, de 27 de octubre, sobre extracción y trasplante de órganos, las referencias a la utilización terapéutica de los tejidos humanos de la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios y la ya mencionada Ley 14/2006, de 26 de mayo, sobre técnicas de reproducción humana asistida, en lo que se refiere a la regulación de las células reproductoras. Por último, desde el punto de vista organizativo, también se han tenido en cuenta las previsiones del Real Decreto 2070/1999, de 30 de diciembre, por el que se regulan las actividades de obtención y utilización clínica de órganos humanos y la coordinación territorial en materia de donación y trasplante de órganos y tejidos.
Este real decreto ha sido debatido con numerosos expertos, centros, entidades, corporaciones profesionales y sociedades científicas, así como con otros organismos relacionados con la materia. Asimismo, se ha tratado con los representantes de las comunidades autónomas en la Comisión de Trasplantes y Medicina Regenerativa del Consejo Interterritorial del Sistema Nacional de Salud y se ha sometido al Pleno de dicho Consejo. Se han recabado, además, los informes de la Comisión de seguimiento y control de la donación y utilización de células y tejidos humanos y de la Agencia de Protección de Datos.
Este real decreto, en cuanto determina aspectos esenciales para la protección de la salud y de la seguridad de las personas, tanto de los donantes como de los posibles receptores, tiene la condición de normativa básica, de acuerdo con lo previsto en la Ley 14/1986, de 25 de abril, General de Sanidad, y al amparo del artículo 149.1.16.ª de la Constitución.
En su virtud, a propuesta de la Ministra de Sanidad y Consumo, previa aprobación del Ministro de Administraciones Públicas, de acuerdo con el Consejo de Estado y previa deliberación del Consejo de Ministros en su reunión del día 10 de noviembre de 2006,
DISPONGO:
1. Este real decreto regula las actividades relacionadas con la utilización de células y tejidos humanos y los productos elaborados derivados de ellos, cuando están destinados a ser aplicados en el ser humano. Las actividades reguladas incluyen su donación, obtención, evaluación, procesamiento, preservación, almacenamiento, distribución, aplicación e investigación clínica.
2. En el caso de que la elaboración, transformación, procesamiento, aplicación e investigación clínica de los productos derivados de las células y tejidos estén regulados por normas específicas, este real decreto sólo se aplicará a su donación, obtención y evaluación.
3. Quedan excluidos del ámbito de este real decreto:
a) Las células y tejidos utilizados como injertos autólogos dentro del mismo proceso quirúrgico.
b) La sangre, los componentes y los derivados sanguíneos tal y como se definen en el Real Decreto 1088/2005, de 16 de septiembre, por el que se establecen los requisitos técnicos y condiciones mínimas de la hemodonación y de los centros y servicios de transfusión.
c) Los órganos o partes de órganos, si su fin es el de ser utilizados en el cuerpo humano con la misma función que el órgano completo.
4. Este real decreto se aplicará a las células reproductoras en todo lo no previsto en la Ley 14/2006, de 26 de mayo, sobre técnicas de reproducción humana asistida, y en su normativa de desarrollo.
1. A efectos de este real decreto se entenderá por:
a) Almacenamiento: mantenimiento de las células o tejidos bajo condiciones controladas y apropiadas hasta su distribución.
b) Aplicación: cualquier actividad que implique el uso de células o tejidos en un receptor humano y/o en aplicaciones extracorporales (se engloban las actividades de implantar, infundir, injertar, aplicar o trasplantar).
c) Células: las células individuales de origen humano o los grupos celulares de origen humano cuando no estén unidos por ninguna forma de tejido conectivo.
d) Células reproductoras: aquellas células o tejidos que puedan ser utilizados para la reproducción humana asistida.
e) Centro o unidad de obtención: establecimiento sanitario, unidad hospitalaria o cualquier otra institución que lleve a cabo actividades de obtención y extracción de tejidos o células, o que puede posibilitar la recogida y utilización de residuos quirúrgicos con las finalidades que establece esta norma, y que no precise ser autorizado como establecimiento de tejidos.
f) Centro o unidad de implante o aplicación en humanos: establecimiento sanitario, unidad hospitalaria o cualquier otra institución que lleve a cabo actividades de aplicación de células o tejidos humanos en humanos.
g) Cuarentena: periodo en el que los tejidos o las células extraídas se mantienen aislados físicamente o por otros métodos efectivos, mientras se espera una decisión sobre su aceptación o rechazo para el uso en humanos.
h) Crítico: hecho, acción o evento que potencialmente puede tener efecto sobre la calidad y la seguridad de las células y tejidos.
i) Distribución: transporte y entrega de tejidos o células destinados a ser aplicados en el ser humano.
j) Donación: el hecho de donar tejidos o células humanos destinados a ser aplicados en el ser humano.
k) Donación dentro de la pareja: la cesión de células reproductoras de un hombre a una mujer de una misma pareja que declaran mantener una relación física íntima.
l) Donante: toda fuente humana, viva o muerta, de células y/o tejidos humanos.
m) Efecto adverso grave: cualquier hecho desfavorable vinculado a la obtención, evaluación, procesamiento, almacenamiento y distribución de células y tejidos que pueda conducir a la transmisión de una enfermedad, a la muerte del paciente, o a estados que hagan peligrar su vida, a minusvalías o incapacidades o que puedan dar lugar a hospitalización o enfermedad o la pueda prolongar.
n) Establecimiento de tejidos: banco de tejidos, unidad de un hospital o cualquier otro centro donde se lleven a cabo actividades de procesamiento, preservación, almacenamiento o distribución de células y tejidos humanos después de su obtención y hasta su utilización o aplicación en humanos. El establecimiento de tejidos también puede estar encargado de la obtención y evaluación de tejidos y células.
ñ) Investigación clínica: investigación desarrollada mediante protocolos que incluyen los procedimientos de obtención y aplicación de células y tejidos humanos en humanos, cuando la eficacia o seguridad de los procedimientos o de las células o tejidos no están suficientemente comprobadas o cuando la indicación terapéutica no está suficientemente consolidada, y cuya finalidad es la comprobación de alguno de estos puntos.
o) Obtención: proceso por el que se puede disponer de células y/o tejidos humanos con la finalidad a que se refiere este real decreto.
p) Órgano: una parte diferenciada y vital del cuerpo humano formada por diferentes tejidos, que mantiene su estructura, vascularización y capacidad para desarrollar funciones fisiológicas con un nivel de autonomía importante.
q) Preservación: utilización de agentes químicos, alteración de las condiciones medioambientales o aplicación de otros medios durante el procesamiento de los tejidos o células, a fin de impedir o retrasar el deterioro biológico o físico de los mismos.
r) Procedimientos operativos estandarizados (POE): instrucciones de trabajo documentadas y autorizadas que describen cómo llevar a cabo actividades o realizar test que habitualmente no se describen en los planes de trabajo o las normas de buenas prácticas.
s) Procesamiento: operación u operaciones que implican la preparación, manipulación, preservación y acondicionamiento de los tejidos y las células destinados a su aplicación en el ser humano.
t) Reacción adversa grave: respuesta inesperada del donante o del receptor, incluida una enfermedad transmisible, asociada a la obtención o aplicación en el ser humano de tejidos y células, que resulte mortal, potencialmente mortal, discapacitante, que produzca invalidez o incapacidad, o que dé lugar a hospitalización o enfermedad o que las prolongue.
u) Sistema de calidad: comprende la estructura orgánica, la definición de responsabilidades, los procedimientos, procesos y recursos, que se destinan a desarrollar la gestión de la calidad. Incluye cualquier actividad que contribuya a la calidad total de forma directa o indirecta.
v) Sistema de gestión de calidad: actividades coordinadas destinadas a la dirección y control de una organización en relación con la calidad.
w) Tejido: toda parte constituyente del cuerpo humano formada por células unidas por algún tipo de tejido conectivo.
x) Trazabilidad: capacidad para ubicar, localizar e identificar las células y/o tejidos en cualquier paso del proceso desde la donación, la obtención, el procesamiento, la evaluación, el almacenamiento y la distribución hasta llegar al receptor o hasta ser desestimados y/o destruidos, lo que lleva consigo la capacidad de identificar al donante, el establecimiento de tejidos y la instalación que recibe, procesa o almacena los tejidos o células, así como la capacidad de identificar al receptor o receptores en los que se apliquen los tejidos o células. La trazabilidad cubre, asimismo, la capacidad de localizar e identificar cualquier dato relevante de los productos y materiales que van a estar en contacto directo con las células y/o tejidos y que puedan afectar a la calidad y seguridad de los mismos.
2. Asimismo, se entenderá por:
a) Uso alogénico: proceso mediante el cual las células o tejidos son extraídos de una persona y aplicados a otra.
b) Uso autólogo: proceso mediante el cual las células o los tejidos son extraídos y aplicados a la misma persona.
c) Uso autólogo eventual: las células y/o tejidos son obtenidos con la finalidad de ser preservados para su aplicación hipotética futura en la misma persona, sin que exista una indicación médica establecida en el momento de la obtención e inicio de la preservación.
d) Uso directo: cualquier procedimiento en el que las células son obtenidas y usadas sin mediar ningún tipo de procesamiento o almacenamiento.
e) Validación: evidencia documental que prueba, con un elevado nivel de garantía, que un determinado proceso, equipo o parte de un equipo o condición ambiental acaba produciendo, de forma consistente y reproducible, un determinado producto que cumple las especificaciones, cualidades y atributos que se habían predeterminado. Un proceso es validado con vistas a probar su efectividad para un uso determinado.
1. La donación de células y tejidos será, en todo caso, voluntaria y altruista, no pudiéndose percibir contraprestación económica o remuneración alguna ni por el donante ni por cualquier otra persona física ni jurídica.
2. Los procedimientos médicos relacionados con la extracción no serán, en ningún caso, gravosos para el donante vivo, ni para la familia en el caso del donante fallecido, debiendo garantizarse al donante vivo la asistencia precisa para su restablecimiento.
3. Los donantes vivos de células o tejidos podrán recibir una compensación de la institución responsable de la extracción, limitada, estrictamente, a cubrir los gastos e inconvenientes derivados de su obtención en concepto de dietas, restitución de ingresos económicos perdidos o similares.
4. No se exigirá al receptor contraprestación alguna por las células y/o tejidos utilizados.
5. Las actividades de los establecimientos de tejidos no tendrán carácter lucrativo, y exclusivamente podrán repercutirse los costes efectivos de los servicios prestados por el desarrollo de las actividades autorizadas.
1. La promoción y publicidad de la donación u obtención de tejidos y células humanos se realizará siempre de forma general, sin buscar un beneficio para personas concretas, y señalándose su carácter voluntario, altruista y desinteresado.
2. La promoción y publicidad de los centros y servicios a que se refiere este real decreto se realizarán así mismo con carácter general y estarán sometidas a la inspección y control de las administraciones sanitarias competentes, conforme establece el artículo 30.1 de la Ley 14/1986, de 25 de abril, General de Sanidad.
3. La existencia y/o persistencia de publicidad y promoción falsa, engañosa o tendenciosa será incompatible con la autorización de actividades de obtención, preservación, procesamiento, distribución o aplicación de células y tejidos en España por parte del centro, institución, unidad o establecimiento de tejidos que haya emitido dicha publicidad o tenga relaciones contractuales con la institución que haya emitido la publicidad.
En particular, se entenderá que existe publicidad engañosa en el caso de los establecimientos, centros, unidades e instituciones cuya publicidad induzca a error sobre la utilidad real de la obtención, procesamiento y preservación de células y tejidos humanos para usos autólogos eventuales, de acuerdo con los conocimientos y experiencia disponibles.
1. Las autoridades sanitarias promoverán la información y la educación de la población general en materia de donación de células y tejidos para su aplicación en humanos, tanto de los beneficios que suponen para las personas que los necesitan como de las condiciones, requisitos y garantías que este procedimiento supone.
2. Asimismo, promoverán la formación continuada de los profesionales sanitarios en esta materia.
1. Se garantizará a los donantes la confidencialidad de todos los datos relacionados con su salud y facilitados al personal autorizado, así como de los resultados y la trazabilidad de sus donaciones, de acuerdo con la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal.
2. Los establecimientos de tejidos deberán adoptar, en el tratamiento de los datos relacionados con los donantes, las medidas de seguridad de nivel alto previstas en el Reglamento de medidas de seguridad de los ficheros automatizados que contengan datos de carácter personal, aprobado por el Real Decreto 994/1999, de 11 de junio.
3. Los datos de carácter personal tendrán carácter confidencial y estarán exclusivamente a disposición de los interesados, conforme a lo dispuesto en la Ley 41/2002, de 14 de noviembre, básica reguladora de la autonomía del paciente y de derechos y obligaciones en materia de información y documentación clínica y, en su caso, de la autoridad judicial para el ejercicio de las funciones que tiene encomendadas. Su utilización se limitará a fines asistenciales o de interés para la salud pública y será recogida y custodiada conforme a lo dispuesto en el artículo 10 de la Ley 14/1986, de 25 de abril, General de Sanidad, en la Ley 15/1999, de 13 de diciembre, y en la citada Ley 41/2002, de 14 de noviembre.
4. El deber de confidencialidad no impedirá la adopción de medidas preventivas cuando se sospeche la existencia de riesgos para la salud individual o colectiva en los términos previstos en los artículos 25 y 26 de la Ley General de Sanidad o, en su caso, conforme a lo que establece la Ley Orgánica 3/1986, de 14 de abril, de Medidas Especiales en Materia de Salud Pública, la Ley 41/2002, de 14 de noviembre, y la Ley Orgánica 15/1999, de 13 de diciembre.
5. No podrán facilitarse ni divulgarse informaciones que permitan la identificación de donantes y receptores de células y tejidos humanos, ni podrán facilitarse a los donantes o sus familiares los datos identificadores de los receptores o viceversa.
1. La obtención de células y tejidos de una persona viva para su ulterior aplicación alogénica en seres humanos podrá realizarse si el donante es mayor de edad, cuenta con plena capacidad de obrar y estado de salud adecuado y ha prestado por escrito su consentimiento informado.
La información que recibirá el donante del médico que haya de realizar la extracción o sea responsable de esta, debe cubrir el objetivo y la naturaleza de la obtención de las células y tejidos; sus consecuencias y riesgos; las pruebas analíticas que se han de realizar; el registro y protección de los datos; y los fines terapéuticos. Asimismo se informará de las medidas de protección aplicables al donante y de los beneficios que con el uso del tejido o grupo celular extraído se espera que haya de conseguir el receptor.
El consentimiento podrá ser revocado en cualquier momento antes de la obtención de la célula y/o el tejido, excepto en los casos de obtención de progenitores hematopoyéticos de sangre periférica o de médula ósea, en que la revocación sólo podrá producirse antes del inicio del tratamiento de acondicionamiento en el receptor.
No podrán obtenerse células y tejidos de personas menores de edad o de personas que por deficiencias psíquicas, enfermedad mental, incapacitación legal o cualquier otra causa, no puedan otorgar su consentimiento, salvo cuando se trate de residuos quirúrgicos o de progenitores hematopoyéticos u otros tejidos o grupos celulares reproducibles cuya indicación terapéutica sea o pueda ser vital para el receptor. En estos casos, el consentimiento será otorgado por quien ostente la representación legal.
2. La obtención de células y tejidos de una persona viva para su procesamiento y posterior uso autólogo o para su uso autólogo eventual se realizará según lo dispuesto en los párrafos primero a tercero del apartado anterior.
En el supuesto de uso autólogo eventual, el contenido de la información facilitada con anterioridad a la obtención deberá incluir, además de lo previsto en el apartado anterior, la indicación de que las células y tejidos así obtenidos estarán a disposición para su uso alogénico en otros pacientes en el caso de existir indicación terapéutica; la información actual, veraz y completa sobre el estado de los conocimientos científicos respecto de los usos terapéuticos o de investigación; las condiciones de procesamiento y almacenamiento en los establecimientos autorizados; y cualquier otra cuestión relacionada con la utilidad terapéutica de la obtención de células y tejidos sin indicación médica establecida en el momento de la obtención e inicio de la preservación.
En el caso de personas menores de edad o de personas que por deficiencias psíquicas, enfermedad mental, incapacitación legal o cualquier otra causa, no puedan otorgar su consentimiento, este será prestado por su representante legal.
3. En todo lo no dispuesto en este artículo, la obtención de células y tejidos de un donante vivo se regirá por lo dispuesto en el capítulo IV de la Ley 41/2002, de 14 de noviembre.
1. La obtención de tejidos y células de personas fallecidas podrá realizarse en el caso de que no hubieran dejado constancia expresa de su oposición, según lo dispuesto en el artículo 11 de la Ley 41/2002, de 14 de noviembre.
En el caso de que se trate de menores o personas incapaces de consentir, la oposición a la donación podrá hacerse constar por quienes hubieran ostentado en vida de aquellos su representación legal.
2. La obtención de material reproductor de personas fallecidas con finalidad reproductiva se regirá por lo dispuesto en la Ley 14/2006, de 26 de mayo, sobre técnicas de reproducción humana asistida.
3. Se deberá facilitar a los familiares y allegados información sobre la necesidad, naturaleza y circunstancias de la obtención, especificando qué procedimientos de restauración y conservación del cadáver y prácticas de sanidad mortuoria se llevarán a cabo.
4. La obtención de células y tejidos se realizará tras la correspondiente certificación de la muerte y la práctica de las diligencias policiales y judiciales si las hubiera.
1. La obtención de tejidos y células podrá realizarse sólo en aquellos centros o unidades sanitarias que estén debidamente autorizados por la autoridad sanitaria competente, según lo dispuesto en el Real Decreto 1277/2003, de 10 de octubre, por el que se establecen las bases generales sobre autorización de centros, servicios y establecimientos sanitarios, y siempre que se cumpla con los requisitos y condiciones mínimas recogidos en el anexo I.1 de este real decreto.
Sin perjuicio de la normativa específica al respecto en cada comunidad autónoma, la solicitud de autorización deberá contener:
a) El nombre del responsable o los responsables del proceso de evaluación del donante y de extracción.
b) Una memoria detallada con la descripción de los medios de que dispone y su adecuación a lo especificado las condiciones y requisitos mínimos.
2. Estos centros y unidades sanitarias deberán contar con una autorización específica para la obtención de cada tipo de tejido o grupo celular, cuya validez se extenderá por un periodo de tiempo determinado no inferior a dos años ni superior a cuatro, al término del cual se podrá proceder a su renovación, previa constatación de que persisten las condiciones y requisitos que dieron lugar a su concesión. En ningún caso se entenderá prorrogada de forma automática.
Cualquier modificación sustancial en las condiciones o requisitos que motivaron la concesión de la autorización deberá ser notificada a la autoridad sanitaria competente, y podrá dar lugar a su revisión o incluso a la revocación de la autorización si las modificaciones suponen una alteración sustancial de las circunstancias que justificaron la concesión.
3. En aquellos casos en los que sea factible y necesaria la obtención del tejido o grupo celular fuera del ámbito hospitalario o sanitario o en un centro sanitario no autorizado para la obtención de tejidos y/o células, dicha obtención deberá ser efectuada por profesionales integrados en un equipo de obtención de un centro debidamente autorizado para tal actividad y en las condiciones que marque dicho centro. En estos supuestos los equipos de obtención de células y tejidos deberán estar en posesión de la debida autorización para esta práctica específica. En todo caso se recogerán los antecedentes clínicos y las muestras necesarias para garantizar que se realicen los estudios y pruebas pertinentes y especificadas en el artículo siguiente.
1. La obtención de tejidos se llevará a cabo de forma que se garantice que la evaluación y selección de los donantes se realiza de acuerdo con los requisitos especificados en los anexos II, III, IV y V de este real decreto, y por personal con la formación y experiencia adecuadas. La persona responsable del procedimiento de selección y evaluación elaborará y firmará el correspondiente informe en el que se recoja el cumplimiento de esos requisitos.
2. La aplicación de criterios de selección y evaluación estará basada en la aplicación de un análisis de la valoración de los riesgos en relación con el uso específico de cada tejido o grupo celular.
3. Los resultados de los procedimientos de selección y evaluación del donante quedarán debidamente documentados y, en su caso, se comunicarán en los términos de la Ley 41/2002, de 14 de noviembre.
4. En el caso de donaciones de múltiples tejidos o de tejidos con finalidad de ser trasplantados de forma no diferida, la unidad responsable del proceso de obtención del centro autorizado se responsabilizará de la custodia y archivo de todos los datos derivados del proceso de selección y evaluación de donantes, así como de la existencia y mantenimiento de la seroteca.
5. En el caso de que las células o tejidos obtenidos vayan a ser enviados a otro establecimiento de tejidos para su procesamiento, este podrá encargarse de completar la evaluación y selección de las células o tejidos, será responsable de determinar su viabilidad final y, además, tendrá acceso a los datos relativos a la evaluación de los donantes y garantizará la custodia de la información sobre la evaluación adicional. De la misma manera, deberá guardar muestras de suero en el caso de haber realizado test adicionales a los procesados en el centro de obtención.
6. En el caso de donaciones para usos específicos diferentes de los trasplantes clínicos, el responsable del procedimiento de obtención se responsabilizará también de las cuestiones relativas a la recogida y archivo de datos y muestras de los donantes.
1. La obtención de las células y de los tejidos deberá realizarse mediante procedimientos operativos estandarizados debidamente documentados y validados que sean adecuados para el tejido o grupo celular a extraer, que en el caso de donantes vivos garanticen su salud y seguridad y respeten su intimidad, y que se ajusten a lo dispuesto en el anexo V.
2. El procedimiento de obtención deberá ser el adecuado para proteger debidamente aquellas propiedades de las células o tejidos que son necesarias para su uso clínico, a la vez que se minimizan los riesgos de contaminación microbiológica.
3. En el caso de que los tejidos y/o células vayan a ser enviados a un establecimiento de tejidos para su procesamiento, el procedimiento de obtención, empaquetado, etiquetado, mantenimiento y transporte hasta dicho centro deberá constar en un documento acordado entre la unidad de obtención y el establecimiento de tejidos.
1. El empaquetado, mantenimiento, etiquetado y transporte de los tejidos y células hasta el establecimiento de tejidos deberán realizarse mediante procedimientos operativos estandarizados debidamente documentados y validados, y se ajustarán a lo dispuesto en el anexo V.
2. El empaquetado y transporte de los tejidos y células debe realizarse de modo que se minimicen los riesgos de contaminación y se prevenga el deterioro de las propiedades biológicas necesarias para su posible uso clínico.
1. Los centros y unidades autorizados para la obtención de células y tejidos deberán disponer de un sistema de recogida y custodia de la información de sus actividades que permita el cumplimiento de las previsiones sobre codificación y trazabilidad de este real decreto.
2. Los centros y unidades autorizados para la obtención de células y/o tejidos deberán facilitar los datos relativos a su actividad que les sean requeridos por las autoridades sanitarias competentes de su comunidad autónoma, que los remitirá a la Organización Nacional de Trasplantes según lo previsto en el capítulo V de este real decreto.
1. Las actividades relacionadas con el procesamiento, almacenamiento y distribución de células y tejidos humanos podrán realizarse sólo en aquellos centros o unidades sanitarias debidamente autorizados por la autoridad sanitaria competente, siguiendo las bases generales de autorización de centros, servicios y establecimientos sanitarios que establece el Real Decreto 1277/2003, de 10 de octubre, y siempre que se cumpla con los requisitos y condiciones mínimas recogidos en el anexo I.2 de este real decreto.
Sin perjuicio de la normativa específica al respecto en cada comunidad autónoma, la solicitud de autorización deberá acompañarse de una memoria en la que se recoja el cumplimiento de los requisitos exigidos en este real decreto.
2. Estos centros y unidades sanitarias deberán contar con una autorización específica para el desarrollo de cada uno de los procesos y actividades del apartado anterior por cada tipo de tejido o grupo celular, según los requisitos recogidos en el anexo I.3. La validez de las autorizaciones se extenderá por un periodo de tiempo determinado no inferior a dos años ni superior a cuatro, al término del cual se podrá proceder a su renovación, previa constatación de que persisten las condiciones y requisitos que dieron lugar a su concesión. En ningún caso se entenderá prorrogada de forma automática.
Cualquier modificación sustancial en las condiciones o requisitos que motivaron la concesión de la autorización deberá ser notificada a la autoridad sanitaria competente, y podrá dar lugar a su revisión o incluso a la revocación de la autorización si las modificaciones suponen una alteración sustancial de las circunstancias que justificaron la concesión.
3. Las solicitudes de autorización de actividades deberán recoger las actuaciones que el establecimiento de tejidos emprenderá en el supuesto de cese de la actividad para la que se solicita la autorización, incluyendo las cobertura de las responsabilidades adquiridas y el envío a otro establecimiento de tejidos debidamente autorizado de las muestras de células y tejidos almacenados, de los sueros y de la información necesaria para asegurar su trazabilidad.
1. Las actividades de procesamiento realizadas en los establecimientos de tejidos tendrán por objeto la preparación, preservación y almacenamiento de las células y tejidos para su uso clínico, tanto autólogo como alogénico, bien en procedimientos terapéuticos con indicaciones médicas establecidas o en procedimientos de aplicación en humanos en casos de utilidad y eficacia debidamente contrastada, o bien en procedimientos de investigación clínica debidamente documentados.
2. Los establecimientos de tejidos procesarán, preservarán y almacenarán las células y tejidos de forma que se garantice su máximo aprovechamiento. Asimismo y según el principio de distribución equitativa, garantizarán el acceso a las células y tejidos en los casos de disponibilidad insuficiente y por razones médicas de idoneidad de los receptores.
3. Según lo previsto en el artículo 3.5, las autoridades competentes de las comunidades autónomas establecerán el régimen de compensación y cargo de los costes que podrá aplicarse a los tejidos y grupos celulares distribuidos para poder cubrir los gastos derivados de su actividad. Estos cargos sólo se podrán aplicar al centro o unidad de aplicación una vez finalizada la actividad de procesamiento o preservación y distribuido el tejido o grupo celular.
4. Los establecimientos de tejidos que preserven células y tejidos para usos autólogos eventuales vienen obligados además a suscribir un seguro que cubra los costes de procesamiento, preservación y almacenamiento para el supuesto de que se produzca la cesión o el envío de esas células y tejidos a otro establecimiento, centro o unidad sanitaria para usos alogénicos en procedimientos terapéuticos con indicaciones médicas establecidas en receptores adecuados. El seguro cubrirá también la cesión en los casos de cese de la actividad del establecimiento.
1. Los establecimientos de tejidos deberán desarrollar y mantener actualizado un sistema de calidad y de gestión de calidad integrado en las directrices y estrategias del establecimiento de tejidos y que incluya como mínimo la siguiente documentación:
a) Manuales de procedimientos operativos de las actividades autorizadas y de los procesos críticos.
b) Manuales de formación y referencia.
c) Formularios de transmisión de la información.
d) Datos relativos al origen y el destino de los grupos celulares o tejidos.
e) Información sobre la trazabilidad de las células o tejidos.
f) Sistema de detección y comunicación de efectos y reacciones adversos.
2. La documentación referida deberá estar disponible para las inspecciones de la autoridad sanitaria competente.
1. Cada establecimiento de tejidos designará a un responsable técnico que deberá reunir las siguientes condiciones:
a) Poseer un título universitario superior en el ámbito de la Medicina o las ciencias biomédicas, expedido tras cursar estudios universitarios completos reconocidos y homologados en España como equivalente a título universitario superior.
b) Tener una experiencia práctica demostrada no inferior a tres años en el ámbito de actuación de que se trate.
2. Entre las funciones y responsabilidades del responsable técnico se incluyen las siguientes:
a) Velar por que en el ámbito del establecimiento de tejidos del que es responsable, los tejidos y células destinados a ser aplicados en humanos se procesen, almacenen y distribuyan de conformidad con lo establecido en este real decreto y en la normativa que resulte de aplicación.
b) Facilitar información a las autoridades competentes sobre las condiciones, requisitos y régimen de funcionamiento exigidos a los establecimientos de tejidos por este real decreto.
c) Aplicar en el establecimiento de tejidos todas las condiciones y requisitos e implantar el régimen de funcionamiento regulados en este real decreto.
3. Los establecimientos de tejidos notificarán a la autoridad competente el nombre y las cualificaciones del responsable técnico. Cuando sea sustituido de forma permanente o transitoria, esta sustitución será comunicada inmediatamente a la autoridad competente. En dicha comunicación deberá incluirse el nombre y cualificación del sustituto y la fecha exacta del periodo de sustitución o de su inicio cuando esta sea indefinida.
4. El personal del establecimiento de tejidos implicado en las actividades relacionadas con el procesamiento, preservación, almacenamiento o distribución de células y tejidos deberá tener la cualificación necesaria para efectuar las tareas que le son encomendadas y recibir la formación pertinente.
1. El establecimiento de tejidos deberá disponer de un procedimiento documentado de recepción que permita verificar que los tejidos y células extraídos en los centros o unidades de obtención cumplen con las exigencias de este real decreto.
2. Los envíos de tejidos y células que no cumplan estas exigencias deben ser rechazados por el establecimiento de tejidos.
3. La recepción de los tejidos y células debe ajustarse a lo dispuesto en los anexos V.2 y VI.
4. El proceso de recepción debe asegurar que no existe riesgo de contaminación con los tejidos y células ya depositados y que estén en fase de procesamiento, preservación o almacenamiento.
1. El establecimiento de tejidos incluirá en sus manuales de procedimiento toda actividad de procesamiento de las células y tejidos, y velará porque se lleven a cabo en condiciones controladas. Se verificará que el equipo utilizado, el entorno de trabajo y la concepción, validación y condiciones de control de los procesos se ajusten a los requisitos que se especifican en el anexo I.3.
2. Cualquier modificación de los procesos utilizados en la preparación de los tejidos o células deberá cumplir los mencionados requisitos.
1. Cualquier actuación relacionada con el almacenamiento de células y tejidos deberá estar documentada en los manuales de procedimientos. Las condiciones de almacenamiento se ajustarán a lo establecido en el anexo I.3 de forma que se garantice el mantenimiento de la viabilidad, calidad y seguridad de las células y tejidos.
2. Según lo dispuesto en el artículo 14.3, en caso de cese de actividad del establecimiento de tejidos, las células y tejidos preservados o almacenados deberán ser transferidos a otro establecimiento de tejidos debidamente autorizado.
Los establecimientos de tejidos deben garantizar la transferencia en caso de cese de la actividad mediante acuerdos previamente establecidos con otros establecimientos y conocidos por las unidades de coordinación de trasplantes de las comunidades autónomas.
3. Todas las informaciones sobre las actividades de almacenamiento serán debidamente recogidas y custodiadas con el fin de que pueda conocerse en todo momento la situación de disponibilidad de las células y tejidos almacenados.
Los procedimientos de etiquetado, documentación y acondicionamiento se ajustarán a lo establecido en el anexo I.3.
1. Las condiciones de distribución y transporte de los tejidos y células se ajustarán a lo dispuesto en el anexo I.3.
2. El transporte desde el establecimiento de tejidos hasta el centro de implante o hasta otro establecimiento de tejidos se realizará por los medios más adecuados de transporte terrestre o aéreo y a través de sistemas capaces de mantener la viabilidad y funcionalidad de las células y/o tejidos. Estos sistemas deberán especificarse en procedimientos documentados según el tipo de célula o tejido a trasladar.
1. El Ministerio de Sanidad y Consumo autorizará, previo informe de la Organización Nacional de Trasplantes, la importación y exportación de los tejidos y células a los que se refiere este real decreto. La importación, exportación y tránsito de estas células y tejidos sólo se efectuará a través de los recintos aduaneros especificados en el anexo I del Real Decreto 65/2006, de 30 de enero, por el que se establecen requisitos para la importación y exportación de muestras biológicas.
2. Sólo se autorizará la importación de tejidos y células si concurren las siguientes circunstancias:
a) Que existe un probado beneficio en la utilización de los tejidos y células que se pretenden aplicar.
b) Que la finalidad de los tejidos y/o células es la de su aplicación en humanos.
c) Que, en el caso de tratarse de células y tejidos que habitualmente se procesan en alguno de los establecimientos de tejidos nacionales, no existe, en ese momento, disponibilidad de dichas células y/o tejidos.
3. Sólo se autorizará la exportación de tejidos y células si concurren las siguientes circunstancias:
a) Que existe disponibilidad suficiente de dichas células y/o tejidos en los establecimientos de tejidos nacionales.
b) Que existe una razón médica que justifique la exportación.
4. Las solicitudes de importación y exportación de células y tejidos se presentarán en la Organización Nacional de Trasplantes por el establecimiento de tejidos, el centro o la unidad implicada, con el conocimiento previo de la unidad de coordinación de trasplantes de la comunidad autónoma que corresponda. La Organización Nacional de Trasplantes dará traslado de las solicitudes a la Subdirección General de Sanidad Exterior del Ministerio de Sanidad y Consumo junto con su informe para su tramitación.
5. En las solicitudes de importación y exportación de células y tejidos se especificará la institución de origen y destino, respectivamente, que deben cumplir normas de calidad y de seguridad equivalentes a las reguladas en este real decreto.
6. Con el fin de asegurar el cumplimiento de lo previsto en el apartado anterior, el establecimiento de tejidos expedirá un certificado que acompañará a la solicitud de importación y exportación. En el caso de las importaciones de tejidos y células el certificado deberá contener la siguiente información:
a) Un informe técnico documentado en el que conste que el tejido las células o la forma en que se han procesado, son imprescindibles para el procedimiento terapéutico que se va a aplicar y que, o bien los tejidos y/o células, o bien el método de procesamiento, no están disponibles ni pueden ser proporcionados por los establecimientos nacionales.
b) La documentación relativa a la institución de origen donde consten las garantías éticas y sanitarias que se observan.
c) Una memoria del establecimiento de tejidos de origen donde figuren las evaluaciones y estudios realizados (clínicos, biológicos, microbiológicos y/o inmunológicos), en consonancia con lo establecido en este real decreto respecto de la selección y evaluación del donante.
En el caso de las exportaciones de tejidos y células el certificado deberá contener la siguiente información:
a) Un informe donde conste la suficiente disponibilidad nacional de los tejidos y/o células que se pretenden exportar.
b) La documentación que acredite la no disponibilidad del método de procesamiento a utilizar cuando este sea el motivo de la salida de los tejidos y/o las células.
c) Una memoria técnica donde figuren las razones médicas que justifiquen la salida de los tejidos y/o células cuando éste sea el motivo.
d) La documentación que acredite que se garantiza la protección de los datos.
7. La importación de células o tejidos podrá ser denegada o revocada cuando no procedan de donaciones altruistas realizadas en países terceros que reúnan las debidas garantías.
1. Los establecimientos de tejidos deberán celebrar contratos por escrito con terceros siempre que estos desarrollen una actividad que influya o pueda influir en la calidad y en la seguridad de los tejidos y/o células procesadas, y en particular cuando:
a) El establecimiento de tejidos confíe a un tercero la responsabilidad de una fase del procesamiento de células y/o tejidos.
b) Un tercero suministre materiales o productos o bien preste servicios que puedan afectar a la calidad y seguridad de las células y/o tejidos.
c) Un establecimiento de tejidos preste un servicio a otro establecimiento para el cual no está autorizado.
d) Un establecimiento de tejidos almacene y distribuya tejidos y/o células procesadas o tratadas por un tercero.
2. El establecimiento de tejidos evaluará la capacidad de los terceros y seleccionará a quienes garanticen el cumplimiento las normas establecidas en este real decreto.
3. Los contratos deberán especificar claramente las responsabilidades de los terceros en relación con los procesos que van a llevar a cabo así como una descripción detallada de dichos procesos.
4. Existirán procedimientos operativos documentados donde se especifiquen la forma de contratar, las relaciones entre las partes contratantes y los protocolos que cada uno debe seguir en relación con la actividad contratada.
5. Los establecimientos de tejidos deberán contar con un registro de los contratos celebrados con terceros cuya información estará disponible para la autoridad competente y la unidad de coordinación de trasplantes de la comunidad autónoma correspondiente.
6. En caso de resolución del contrato, la entidad contratada deberá remitir al establecimiento de tejidos los datos y muestras que pueden afectar a la trazabilidad o a la calidad y seguridad de células y tejidos. Los términos de esta remisión de muestras e información deberán detallarse en el procedimiento de contratación y deberán figurar en el contrato de servicio.
7. Los establecimientos de tejidos enviarán copias de los contratos suscritos con terceros a la unidad de coordinación de trasplantes y a la autoridad competente para la autorización de estas actividades de su comunidad autónoma.
8. Cuando la contratación del tercero implique el acceso por parte de éste a datos de carácter personal, el contrato deberá cumplir lo establecido en el artículo 12 de la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal.
1. Los establecimientos de tejidos dispondrán de un sistema de recogida y custodia de la información relativa a sus actividades que asegure la trazabilidad de todas las células y tejidos procesados. En el caso de que el sistema tenga formato electrónico, debe asegurarse la existencia de copias de seguridad.
2. Existirá un procedimiento documentado para la recogida y custodia de la información. El establecimiento designará a una persona como responsable del sistema de recogida y custodia de la información de las actividades y comunicará esta designación a la unidad de coordinación de trasplantes y a la autoridad competente de la comunidad autónoma en la que esté ubicado.
3. Los establecimientos de tejidos remitirán información trimestral de sus actividades a la unidad de coordinación de trasplantes y a la autoridad competente de la comunidad autónoma correspondiente y en todo momento tendrán a disposición de ésta su sistema de recogida y custodia de la información.
1. La aplicación de células y tejidos humanos podrá realizarse sólo en aquellos centros o unidades sanitarias debidamente autorizados por la autoridad sanitaria competente siguiendo las bases generales de autorización de centros, servicios y establecimientos sanitarios que establece el Real Decreto 1277/2003, de 10 de octubre, y siempre que se cumpla con los requisitos y condiciones mínimas recogidos en el anexo I.4 de este real decreto.
2. Estos centros y unidades sanitarias deberán contar con una autorización específica para cada actividad de aplicación o implante de células y tejidos y para cada tipo de células y tejidos. La autoridad sanitaria competente de cada comunidad autónoma determinará el periodo de vigencia de las autorizaciones, que no deberá ser inferior a dos años ni superior a cuatro, así como los requisitos para su posible renovación.
Cualquier modificación sustancial en las condiciones o requisitos que motivaron la concesión de la autorización deberá ser notificada a la autoridad sanitaria competente, y podrá dar lugar a su revisión o incluso a la revocación de la autorización si las modificaciones suponen una alteración sustancial de las circunstancias que justificaron la concesión.
3. Sin perjuicio de la normativa específica al respecto en cada comunidad autónoma, la solicitud de autorización de la aplicación se acompañará de una memoria con la descripción detallada de los medios de que dispone el centro para realizar la actividad solicitada y su adecuación a lo dispuesto en el real decreto. Así mismo, se harán constar el tipo de tejido o grupo de células para la que se solicita la autorización y el nombre y formación de la persona responsable del equipo de implantación.
4. La autoridad sanitaria competente de las comunidades autónomas notificará en tiempo real a la Organización Nacional de Trasplantes las autorizaciones que se concedan, denieguen y revoquen.
5. Para la aplicación de células y tejidos humanos se requerirá el consentimiento del receptor o de sus representantes legales según lo dispuesto en la Ley 41/2002, de 14 de noviembre.
1. Las células y tejidos almacenados en los establecimientos de tejidos estarán a disposición de los centros o unidades de aplicación de tejidos y células para usos alogénicos en procedimientos terapéuticos con indicaciones médicas establecidas en receptores adecuados.
En el caso de que el establecimiento de tejidos que ha procesado y almacenado las células y tejidos no disponga de la necesaria infraestructura para una completa tipificación de las células y tejidos que permita establecer compatibilidades e idoneidades cuando sea preciso, deberá enviar una muestra a otro establecimiento debidamente autorizado que sí esté dotado de la infraestructura adecuada y que se constituirá en establecimiento de referencia. En la distribución de células y tejidos se tendrá en cuenta lo previsto en este real decreto.
2. La aplicación autóloga quedará encuadrada en el caso de procedimientos terapéuticos de eficacia demostrada en indicaciones médicas establecidas.
En el caso de que se realicen actividades de procesamiento para usos autólogos eventuales de los que no hay indicación médica establecida actual, las células y tejidos así procesados estarán disponibles para su aplicación alogénica según lo dispuesto en el apartado primero.
3. En el caso de tratarse de un tejido o grupo celular de limitada disponibilidad, se centralizarán los datos de los pacientes a la espera de recibir el implante en la unidad de coordinación de trasplantes de la comunidad autónoma y en la Organización Nacional de Trasplantes.
4. La solicitud del tejido o grupo celular la efectuará el responsable del centro o la unidad de aplicación al responsable del establecimiento de tejidos. Deberá adjuntarse a la solicitud una copia validada de la autorización como centro o unidad de aplicación de dicho tejido o grupo celular. El establecimiento de tejidos no distribuirá el tejido o grupo celular si no se aporta la copia mencionada.
5. En ausencia de establecimientos de procesamiento de tejidos en la propia comunidad autónoma, o en caso de carecer los establecimientos autorizados del tejido solicitado, la petición se dirigirá a la unidad de coordinación de trasplantes de esa comunidad autónoma quien la remitirá a la Organización Nacional de Trasplantes para su búsqueda a nivel nacional o internacional.
1. Los centros y unidades autorizados para la aplicación en humanos de células o tejidos humanos deberán disponer de un sistema de recogida y custodia de información sobre las actividades realizadas en este ámbito, de acceso restringido y confidencial, donde constarán los usos y aplicaciones clínicos realizados con los datos necesarios para la identificación de los receptores, de los tejidos y/o células implantados así como su procedencia, de forma que se permita el adecuado seguimiento en caso necesario, conforme a lo especificado en el capítulo V.
2. Los centros y unidades autorizados para la aplicación de células o tejidos humanos remitirán información trimestral de sus actividades a la unidad de coordinación de trasplantes y a la autoridad competente de la comunidad autónoma correspondiente y en todo momento tendrán a disposición de ésta su sistema de recogida y custodia de la información.
3. Los centros de aplicación de células y tejidos deberán informar al establecimiento de tejidos o, en su caso, al centro de obtención que les ha suministrado las células y tejidos, sobre el destino final de la aplicación en humanos de dichas células o tejidos, y en el caso de que finalmente no se produzca la aplicación, la causa que no la hizo posible.
1. La investigación clínica con células y/o tejidos sólo podrá llevarse a cabo en los centros y unidades de obtención y aplicación y en los establecimientos de tejidos debidamente autorizados para el desarrollo de la actividad investigadora.
2. Los proyectos de investigación clínica serán autorizados por la autoridad competente de la comunidad autónoma correspondiente. Para la concesión de la autorización será preceptivo el informe de los expertos designados a estos efectos por la Comisión de Trasplantes y Medicina Regenerativa del Consejo Interterritorial del Sistema Nacional de Salud.
3. Las solicitudes de autorización para proyectos de investigación clínica con células y/o tejidos deberán incluir, al menos, la siguiente información y documentación:
a) Justificación y descripción detallada del proyecto de investigación clínica.
b) La información sobre procedimientos de investigación clínica o básica relacionados e información sobre los tejidos/grupos celulares que se van a utilizar y del proceso de procesamiento y/o transformación y utilización de los mismos.
c) Designación del centro coordinador y profesional responsable del proyecto que actúa como investigador principal y descripción del equipo o equipos de investigación.
d) Identificación de los centros y unidades participantes, tanto en la fase de extracción como en la de implante.
e) Identificación de los establecimientos de tejidos cuando sean diferentes de los centros de extracción o implante.
f) Las autorizaciones de los responsables de los centros implicados.
g) El informe del comité de ética del centro coordinador del proyecto. En caso de no ser un centro de implante se requerirá el informe de los comités de ética de los centros de implante implicados.
h) El documento de consentimiento informado.
i) La póliza de contratación de los seguros para los pacientes cuando proceda.
j) El Informe de los costes del proyecto y del organismo promotor.
k) El protocolo del sistema de garantía de calidad del proyecto.
4. La autoridad competente de cada comunidad autónoma deberá notificar cada seis meses a la Organización Nacional de Trasplantes aquellos proyectos de investigación clínica que se encuentran autorizados y en ejecución en el ámbito de su comunidad autónoma.
5. Lo previsto en este artículo no será aplicable a los supuestos de investigación clínica en terapia celular, que se regularán según lo dispuesto en el Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos. En estos casos y con carácter preceptivo, la Agencia Española de Medicamentos y Productos Sanitarios solicitará informe a la Organización Nacional de Trasplantes.
1. La Organización Nacional de Trasplantes, sin perjuicio de las competencias de registro de las autoridades autonómicas, desarrollará y mantendrá un registro de establecimientos de tejidos y de unidades o centros de obtención y aplicación de células y tejidos humanos autorizados, donde se especificarán para cada uno de ellos las actividades concretas para las cuales están autorizados. Este registro estará accesible al público.
2. Las unidades de coordinación de trasplantes de las comunidades autónomas deberán comunicar en tiempo real a la Organización Nacional de Trasplantes la información relativa a los establecimientos de tejidos y centros o unidades de obtención y aplicación de tejidos y células que se autoricen en el ámbito de su competencia, con el fin de incluirla en este registro. Dicha información deberá incluir, al menos, el nombre y ubicación del establecimiento, unidad o centro autorizado, las actividades para las que están autorizados y los periodos de vigencia de dichas autorizaciones.
3. La Organización Nacional de Trasplantes designará un responsable técnico del mantenimiento y custodia del registro.
1. Las autoridades competentes de las comunidades autónomas determinarán la información requerida según lo previsto en los artículos 13, 25 y 28 de este real decreto, que al menos incluirá los contenidos mínimos aprobados por la Comisión de Transplantes y Medicina Regenerativa del Consejo Interterritorial del Sistema Nacional de Salud.
2. Las unidades de coordinación de trasplantes o, en su caso, las autoridades competentes de las comunidades autónomas, enviarán a la Organización Nacional de Trasplantes con, al menos, periodicidad semestral, la información recogida en aplicación de los artículos 13, 25 y 28 de este real decreto.
La Organización Nacional de Trasplantes desarrollará y mantendrá un sistema de recogida, custodia y análisis de dicha información, al que tendrán acceso las unidades de coordinación de trasplantes de las comunidades autónomas en los términos que se acuerden en la Comisión de Transplantes y Medicina Regenerativa del Consejo Interterritorial del Sistema Nacional de Salud.
3. La Organización Nacional de Trasplantes elaborará un informe anual donde figuren las informaciones relativas a los establecimientos de tejidos, unidades o centros de obtención y aplicación de células y tejidos humanos, así como las actividades desarrolladas. Este informe, que en ningún caso contendrá datos personales referidos a los donantes y los receptores, será accesible al público y se remitirá a todos los centros y unidades implicados e incluirá datos de interés general a los que se dará la debida difusión.
4. El Ministerio de Sanidad y Consumo, a través de la Organización Nacional de Trasplantes, colaborará con la Comisión Europea y los demás Estados miembros de la Unión Europea en el desarrollo de una red de intercambio de información entre los registros nacionales de establecimientos de tejidos y de centros o unidades de obtención y aplicación de células y tejidos humanos autorizados.
5. El acceso a cualquiera de los datos contenidos en los sistemas de información regulados en este real decreto quedará restringido a aquellas personas autorizadas tanto por los responsables técnicos de los establecimientos de tejidos y los responsables de las unidades de extracción o de implante de tejidos, como por las unidades de coordinación de trasplantes o las autoridades competentes de la comunidades autónomas y, en el ámbito de su competencia, por la Organización Nacional de Trasplantes.
6. Todos los sistemas de recogida y archivo de información deben cumplir con los principios establecidos en la Ley Orgánica 15/1999, de 13 de diciembre.
1. La Organización Nacional de Trasplantes y las unidades de coordinación de trasplantes de las comunidades autónomas establecerán, en los términos que se acuerden en la Comisión de Trasplantes y Medicina Regenerativa del Consejo Interterritorial del Sistema Nacional de Salud, un sistema de rastreo de origen a destino de todas aquellas células y tejidos humanos obtenidos con el fin de ser aplicados en humanos. Dicho sistema recogerá la información referida en el anexo VI.
2. En el caso de células embrionarias de eventual aplicación en humanos, la Organización Nacional de Trasplantes y los responsables del Banco Nacional de Líneas Celulares y de la Comisión de Seguimiento y Control de la Donación y utilización de Células y Tejidos Humanos, establecerán un sistema que garantice el seguimiento previsto en el apartado anterior.
3. La información, cuando proceda, se codificará con acuerdo a los estándares básicos regulados en los anexos VI y VII lo que permitirá su seguimiento uniforme. Los establecimientos de tejidos y las unidades y centros de obtención y aplicación de células y tejidos deberán recoger la información en tiempo real.
4. Los establecimientos de tejidos recogerán la información del destino de las células y tejidos distribuidos para aplicación en humanos. Dicha información deberá ser facilitada por los centros, organismos o unidades de aplicación de tejidos y células para cada caso en particular, con el fin de asegurar la trazabilidad de las células y tejidos.
5. El rastreo de origen a destino se aplicará no sólo a los productos celulares y tejidos, sino también a los productos y materiales que entren en contacto con dichas células y tejidos y puedan tener efecto sobre su calidad y seguridad.
6. La información se guardará y custodiará de forma segura durante al menos 30 años a partir de su codificación.
1. La Comisión de Transplantes y Medicina Regenerativa del Consejo Interterritorial del Sistema Nacional de Salud, a propuesta de la Organización Nacional de Trasplantes, establecerá un sistema único y obligatorio de codificación, compatible con los sistemas de otros Estados miembros de la Unión Europea, que permitirá identificar de forma única e inequívoca los tejidos y células obtenidos, procesados y distribuidos para su aplicación en humanos.
2. El diseño del sistema de codificación se ajustará a los requisitos mínimos exigidos en el anexo VII.
3. Se desarrollará un sistema técnico que soporte el sistema de codificación y que será accesible para todos los centros y establecimientos autorizados para la obtención, procesamiento y aplicación de células y tejidos, así como para las unidades de coordinación de trasplantes de las comunidades autónomas y la Organización Nacional de Trasplantes.
1. Desde la entrada en vigor de este real decreto funcionará un sistema de biovigilancia que permitirá notificar, registrar y transmitir información sobre los efectos y reacciones adversas graves que puedan haber influido o pudieran influir en la calidad y seguridad de las células y tejidos y que puedan atribuirse a los procesos de obtención, evaluación, procesamiento, almacenamiento y distribución de las células y tejidos, así como toda reacción adversa grave observada durante o a raíz de la aplicación clínica de las células y/o tejidos, y que pudiera estar relacionada con su calidad y seguridad.
2. En tanto no se regule de forma distinta, la red de coordinación de trasplantes de las comunidades autónomas y de la Administración General del Estado funcionará como red de biovigilancia.
3. Todos los centros o unidades que obtengan y apliquen células o tejidos así como los establecimientos de tejidos deberán comunicar la existencia de cualquier evento o reacción adversa en la forma y en los términos establecidos en el anexo VIII, a través de la mencionada red de coordinación de trasplantes.
4. Los establecimientos de tejidos que procesen o preserven tejidos que puedan verse afectados por alguna reacción o efecto adverso grave deberán emitir un informe detallado de las posibles causas y de las consecuencias, así como de las medidas adoptadas y de las que se vayan a adoptar.
5. La Organización Nacional de Trasplantes es responsable de la comunicación de la existencia de efectos adversos graves que pudieran afectar a otros Estados miembros a través del sistema de notificación que establezca la Comisión Europea. Asimismo, notificará a las unidades autonómicas de coordinación de trasplantes donde se ubiquen los establecimientos de tejidos afectados o que pudieran estar afectados por un efecto adverso grave ocurrido en otro país, toda la información relativa a dicho evento.
6. Los establecimientos de tejidos son responsables de garantizar que existe un procedimiento rápido, preciso y verificable que permita retirar de la distribución todo producto que pueda estar relacionado con un efecto adverso grave.
1. Las autoridades competentes de las comunidades autónomas efectuarán inspecciones periódicas para garantizar que los establecimientos de tejidos autorizados en el ámbito de sus competencias cumplen los requisitos de este real decreto y aplican las medidas de control de calidad exigidas en él.
2. El Consejo Interterritorial del Sistema Nacional de Salud, a través de su Comisión de Trasplantes y Medicina Regenerativa, aprobará un plan de inspecciones a iniciativa de la Organización Nacional de Trasplantes y las unidades de coordinación de trasplantes de las comunidades autónomas, en el que se contemplarán las inspecciones periódicas previstas en el apartado anterior.
3. La Comisión de Trasplantes y Medicina Regenerativa elevará para informe del Consejo Interterritorial del Sistema Nacional de Salud los criterios generales que aseguren que las condiciones de realización de las inspecciones, las medidas de control y la formación y la cualificación de los profesionales encargados de ellas, se realizan con un nivel mínimo y homogéneo de competencia y resultados.
4. El intervalo entre dos inspecciones regulares será de dos años.
5. Las autoridades competentes de las comunidades autónomas organizarán las inspecciones extraordinarias y la aplicación de las medidas de control que consideren necesarias ante un efecto o reacción adversa grave. Asimismo, organizarán inspecciones extraordinarias y aplicarán medidas de control en caso necesario, a petición justificada de la autoridad competente de otro Estado miembro de la Unión Europea, o de la Comisión.
6. La inspección no sólo afectará a los establecimientos de tejidos sino también a todos aquellos terceros con los que existan relaciones contractuales, e implicará el examen, evaluación y verificación de cualquier infraestructura, equipamiento, información, documento o registro relacionado con lo regulado en este real decreto.
7. Las peticiones de inspección extraordinaria de otro Estado miembro o de la Comisión deberán canalizarse a través de la Organización Nacional de Trasplantes, quien será así mismo responsable de trasladar al Estado peticionario o a la Comisión el informe con el resultado de la inspección y las medidas de control aplicadas.
1. La autoridad competente de cada comunidad autónoma llevará a cabo los programas de evaluación y acreditación de los centros y servicios de obtención, procesamiento, distribución e implante de células y tejidos de acuerdo con los criterios a las que se hace referencia en el apartado siguiente.
2. La Comisión de Trasplantes y Medicina Regenerativa elevará para informe del Consejo Interterritorial del Sistema Nacional de Salud los criterios generales sobre las condiciones de evaluación y acreditación de centros y servicios.
3. La autoridad competente de cada comunidad autónoma informará periódicamente a la Organización Nacional de Trasplantes, con frecuencia al menos anual, sobre la actividad de evaluación y acreditación de los centros y servicios y sus resultados.
4. Según lo previsto en el artículo 70.2.d) de la Ley 14/1986, de 25 de abril, General de Sanidad, la Organización Nacional de Trasplantes, previo acuerdo de la Comisión de Trasplantes y Medicina Regenerativa del Consejo Interterritorial del Sistema Nacional de Salud, podrá actuar como entidad técnica para la evaluación y acreditación de los centros y servicios autorizados al amparo de lo establecido en este real decreto.
Sin perjuicio de otra normativa que pudiera resultar de aplicación, las infracciones cometidas contra lo dispuesto en este real decreto y sus disposiciones de desarrollo tendrán la consideración de infracción en materia de sanidad, según lo previsto en el capítulo VI del Título I de la Ley 14/1986, de 25 de abril, General de Sanidad, y en las demás disposiciones que resulten de aplicación.
En las infracciones en materia de utilización de ficheros que contengan datos personales se estará a lo dispuesto en el Título VII de la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal.
La Organización Nacional de Trasplantes desarrollará, en un plazo no superior a seis meses desde la entrada en vigor de este real decreto, el sistema de transmisión de la información previsto en el artículo 30.2, que incluirá las herramientas técnicas necesarias para proteger dicha información contra una destrucción accidental o ilícita, o la alteración, difusión o acceso no autorizado. Una vez desarrollado e implantado dicho sistema, las unidades de coordinación de transplantes de las comunidades autónomas procederán al envío de la información en los términos previstos en el artículo mencionado.
Los centros y establecimientos que a la entrada en vigor de este real decreto se encuentren autorizados para realizar las actividades reguladas en el mismo dispondrán de un plazo de 12 meses desde su entrada en vigor para adecuarse a sus disposiciones.
En el caso de no haberse producido la adaptación en este plazo, las autorizaciones de estos centros y establecimientos se entenderán revocadas, sin perjuicio de las sanciones en que pudiesen incurrir en el supuesto de seguir realizando las actividades reguladas por este real decreto.
El Acuerdo Marco de 13 de junio de 1994 suscrito entre el Ministerio de Sanidad y Consumo y la Fundación Internacional «José Carreras» para el trasplante de médula ósea de donantes no emparentados, mantendrá su validez a la entrada en vigor de este real decreto.
El Ministerio de Sanidad y Consumo tomará las medidas necesarias para asegurar que los contenidos que se regulan en dicho Acuerdo se adaptan a las previsiones contenidas en este real decreto.
Quedan derogadas cuantas disposiciones de igual o inferior rango se opongan a lo previsto en este real decreto y, en particular, el Real Decreto 411/1996, de 1 de marzo, por el que se regulan las actividades relativas a la utilización de tejidos humanos.
Este real decreto tiene carácter básico y se dicta al amparo de lo dispuesto en el artículo 149.1.16.ª de la Constitución, que atribuye al Estado la competencia sobre bases y coordinación general de la sanidad. Se exceptúan de lo anterior el artículo 23, que se dicta al amparo de la competencia exclusiva del Estado en materia de sanidad exterior y el artículo 29, que se dicta al amparo de lo dispuesto en el artículo 149.1.15.ª, que atribuye al Estado la competencia exclusiva en materia de fomento y coordinación general de la investigación científica y técnica.
Mediante este real decreto se incorpora al ordenamiento jurídico español la Directiva 2004/23/CE del Parlamento Europeo y del Consejo, de 31 de marzo de 2004, relativa al establecimiento de normas de calidad y de seguridad para la donación, la obtención, la evaluación, el procesamiento, la preservación, el almacenamiento y la distribución de células y tejidos humanos, así como la Directiva 2006/17/CE de la Comisión, de 8 de febrero de 2006, por la que se aplica la Directiva 2004/23/CE del Parlamento Europeo y del Consejo en lo relativo a determinados requisitos técnicos para la donación, la obtención y la evaluación de células y tejidos humanos.
Se habilita al Ministro de Sanidad y Consumo para dictar las disposiciones necesarias para el desarrollo y aplicación de este real decreto, así como para la modificación de sus anexos, con el fin de adecuarlos al avance de los conocimientos científicos y técnicos o para adaptarlos a la normativa comunitaria.
El presente real decreto entrará en vigor el día siguiente al de su publicación en el «Boletín Oficial del Estado».
Dado en Madrid, el 10 de noviembre de 2006.
JUAN CARLOS R.
La Ministra de Sanidad y Consumo,
ELENA SALGADO MÉNDEZ
1. Los requisitos y condiciones mínimas para la autorización de centros sanitarios para obtener células y tejidos humanos para su uso en humanos son:
a) Disponer de una unidad médico quirúrgica especializada en la práctica de la extracción del tejido o grupo celular que se pretenda obtener. Se designará la persona responsable del procedimiento de extracción en cada caso.
b) En el caso de que el uso o destino de las células o tejidos sea un trasplante inmediato o no diferido, deberá existir una relación establecida con el equipo de coordinación de trasplantes.
c) Cuando el destino de las células o tejidos extraídos sea su derivación a un establecimiento de tejidos para su procesamiento, deberá existir un acuerdo de colaboración que incluirá un protocolo consensuado con dicho establecimiento, en el que figuren las condiciones de obtención, preparación y transporte de los tejidos o células hasta su llegada al establecimiento de procesamiento.
d) Cuando el destino de las células o tejidos sea su transformación deberá haber un convenio de colaboración con la entidad responsable de dicha transformación conocido y autorizado por la autoridad competente de la correspondiente comunidad autónoma.
e) Disponer de la infraestructura y personal adecuados para la correcta evaluación del donante y garantizar la realización de los estudios pertinentes para descartar la presencia de enfermedades transmisibles, según lo estipulado en los anexos II, III y IV. Se designará la persona responsable de los procedimientos de evaluación del donante.
f) Disponer de procedimiento operativo estandarizado para la correcta verificación de:
1.º La identidad del donante.
2.º Los requerimientos de la autorización.
3.º Los criterios de selección y evaluación.
4.º La realización de los tests de laboratorio requeridos para la evaluación y selección.
g) Disponer de las instalaciones y medios necesarios para garantizar las condiciones de extracción, preparación y transporte de las células o tejidos, según el protocolo referido en el apartado c).
h) Disponer de las instalaciones y medios adecuados para garantizar la restauración y conservación del cadáver, así como de prácticas mortuorias, en el caso de que la extracción se lleve a cabo de una persona fallecida.
i) Tener establecidas documentalmente las relaciones y condiciones de extracción con los establecimientos de procesamiento y/o implante de tejidos y células con los que se relaciona, que deben ser comunicadas a la autoridad competente.
j) Disponer de un sistema de recogida y custodia de la información relativa a sus actividades, de acceso restringido y confidencial donde constarán las extracciones realizadas y los datos necesarios de los donantes, de las células o tejidos, así como el destino final o intermedio de los mismos. Se conservarán los datos relativos a las pruebas realizadas y características de los donantes, especificándose la fecha de realización y el resultado de las mismas, de forma que se permita el adecuado seguimiento de la información, en caso necesario, conforme a lo previsto en los artículos 13 y 31.
k) Mantener un archivo de sueros de los donantes alogénicos o potencialmente alogénicos (muestras recogidas para un hipotético uso autólogo sin indicación terapéutica actual) durante, al menos, 10 años a partir de la última aplicación clínica o de la fecha de caducidad de las células/tejidos, con el fin de realizar controles biológicos en caso necesario. En el caso de las células reproductoras, el archivo de sueros se mantendrá durante dos años a partir de la última transferencia.
l) Disponer de un procedimiento operativo estandarizado para el envasado, etiquetado y transporte de las células y/o tejidos hasta el punto de destino (establecimientos de tejidos y equipos de trasplante en el caso de distribución directa).
2. Los requisitos técnicos exigidos para optar a la autorización como establecimiento de tejidos son:
a) En cuanto a la organización y dirección:
1.º Además del Director del establecimiento se debe designar un responsable técnico conforme a lo establecido en el artículo 17.
2.º Disponer de una estructura organizativa y régimen de funcionamiento adecuados en los que se definan claramente las relaciones de dependencia y las responsabilidades de cada puesto de trabajo, y que se adecúa a las actividades para los que se solicita la autorización.
3.º Deben existir procedimientos operativos estandarizados de todas las actividades para las que se opta a la autorización.
4.º El establecimiento de tejidos debe dotarse de un equipo médico o tener acceso disponible a un apoyo médico que pueda revisar, supervisar, analizar y, si es necesario, promover determinados cambios en las actividades médicas del establecimiento, como son: selección del donante, evaluación de riesgo de los tejidos/células, disponibilidad de tejidos o células en casos excepcionales, revisión y evaluación del seguimiento de los tejidos/células distribuidos para su aplicación o relaciones con los centros o unidades de aplicación entre otras.
5.º Deben disponer de un sistema de gestión de calidad aplicadas a todas las actividades a cuya autorización opta el Establecimiento de tejidos en las condiciones que marca el artículo 16.
6.º Deben existir procedimientos de minimización de los riesgos inherentes al manejo de material biológico a la vez que se mantiene un adecuado nivel de calidad y seguridad de las células y tejidos. Deben cubrirse en este punto todas las actividades técnicas del procesamiento de células y tejidos del establecimiento, las condiciones ambientales y las condiciones higiénico sanitarias del personal del establecimiento.
7.º Debe existir un procedimiento documentado que asegure la identificación de todos los tejidos y células en cualquier fase de la actividad para la cual se solicita la autorización. Este procedimiento de identificación debe compatibilizarse con el sistema de codificación estatal que se establece en el capítulo VI.
b) En cuanto a personal:
1.º Disponer de personal suficiente y suficientemente preparado para llevar a cabo las actividades para las que solicita la autorización.
2.º Disponer de una descripción detallada del perfil de los puestos de trabajo, tareas, responsabilidades y relaciones con otros puestos de trabajo.
3.º Disponer de un programa de formación continuada para el personal del establecimiento de tejidos que asegure que cada trabajador:
i) Tiene suficiente competencia para el desarrollo de las tareas que le son encomendadas.
ii) Tiene el conocimiento y la experiencia adecuados para entender los procesos científicos técnicos que están en relación con las tareas que le son asignadas.
iii) Conoce la estructura organizativa, el régimen de funcionamiento y el sistema de calidad del establecimiento.
iv) Conoce las normas higiénico-sanitarias del establecimiento en relación con las actividades que allí se llevan a cabo.
v) Está adecuadamente informado de los aspectos éticos y legales y de las normas de correcta práctica en relación con las actividades del establecimiento.
c) En cuanto a equipo y material:
1.º Todo el equipo y el material utilizado debe estar diseñado y mantenido específicamente para el propósito que se persigue, minimizándose cualquier riesgo para los receptores o para el personal que trabaja en el establecimiento.
2.º Los equipos críticos deben ser identificados como tales Se deben inspeccionar de forma periódica y/o regular y se deben adoptar las medidas preventivas adecuadas de acuerdo con las instrucciones del fabricante. Cuando el equipamiento pueda afectar a procesos críticos o a parámetros que marcan las condiciones de almacenamiento o preservación (temperatura, presión, contaje de partículas, niveles de contaminación, etc.) se debe identificar como tal. En estos casos se establecerán los sistemas de monitorización, control y alerta, alarmas y medidas de corrección que sean más adecuadas y requeridas para detectar un mal funcionamiento, el peligro de una desviación inaceptable o un defecto, y se debe asegurar que los parámetros críticos se mantienen en límites aceptables todo el tiempo.
3.º Todo equipamiento nuevo o reparado debe ser evaluado en el momento de su instalación y debe ser validado antes de ser utilizado. La información relativa a todos los resultados de estas evaluaciones debe recogerse y custodiarse.
4.º Las actividades de mantenimiento, limpieza, desinfección, saneamiento y revisión del equipamiento crítico deben figurar en procedimientos documentados, se deben llevar a cabo de forma regular y la información sobre el desarrollo de estos procedimientos se recogerá y custodiará adecuadamente.
5.º Se debe disponer de procedimientos documentados para el funcionamiento de cada uno de los equipos críticos donde se detallarán las acciones que se deben emprender en caso de malfuncionamiento o fallo.
6.º Los procedimientos operativos de las actividades para las que se solicita la autorización deben incluir las especificaciones de los equipos críticos y de los reactivos. Se incluirán las especificaciones de los aditivos (i.e. soluciones) y los materiales de embalaje. Los reactivos y materiales críticos deberán cumplir las especificaciones del Real Decreto 414/1996, de 1 de marzo, por el que ser regulan los productos sanitarios y el Real Decreto 1662/2000, de 29 de septiembre, sobre productos sanitarios para diagnósticos in vitro.
d) En cuanto a infraestructura y locales:
1.º Disponer del local y las infraestructuras necesarias para llevar a cabo las actividades para las que se solicita la autorización.
2.º Cuando estas actividades incluyan el procesamiento de células o tejidos en condiciones de exposición abierta, se deberán especificar las condiciones de calidad de aire y limpieza que exigen para minimizar los riesgos de contaminación incluyendo la contaminación cruzada. La efectividad de las medidas necesarias para cumplir estas condiciones de validarse y monitorizarse.
3.º Excepto en los casos especificados en el punto 4.º, siempre que las células o tejidos se vayan a procesos en exposición abierta y sin un procedimiento posterior de inactivación microbiológica se exigirá una calidad de aire con de partículas y de colonias microbiológicas equivalente a la especificada como grado A en el anexo I de la Guía Europea de Normas de Correcta Fabricación en el lugar del procesamiento. Para el aire ambiente del resto del local de trabajo se exigirá una calidad adecuada para las actividades que se van a llevar a cabo, pero, al menos, equivalente al grado D de la Guía Europea de Normas de Correcta Fabricación en lo que a contaje de partículas y colonias microbiológicas se refiere.
4.º Se podrán admitir unas condiciones medioambientales menos estrictas que las especificadas en el punto 3.º en los siguientes supuestos:
i) Cuando se vayan a aplicar procedimientos de esterilización o inactivación microbiológica validada.
ii) Cuando esté demostrado que la exposición a un aire ambiente de grado A tiene efectos perjudiciales sobre las propiedades biológicas que se requieren al tejido/grupo celular afectado.
iii) Cuando esté demostrado que la vía o procedimiento de aplicación de las células o tejidos implican un riesgo de transmisión de enfermedades bacterianas o fúngicas significativamente menor que el trasplante de células o tejidos.
iv) Cuando no sea técnicamente posible llevar a cabo el procesamiento de las células o tejidos en un ambiente de grado A. Por ejemplo cuando las condiciones de funcionamiento del equipamiento que se requiere no son compatibles con ambiente grado A.
5.º Cuando se aplique alguno de los supuestos del apartado 4.º, se deberán especificar las condiciones del aire ambiente en el que se debe trabajar y se deberá demostrar y documentar que esas condiciones se alcanzan las normas requeridas de calidad y seguridad de los tejidos, teniendo en mente el objeto terapéutico o de aplicación y la vía o modo de aplicación de las células y/o tejidos y la situación inmunológica del receptor. Deberá disponer de la equipación y vestimenta adecuada, tanto para la protección del personal como para la higiene en todos aquellos departamentos donde sea necesario y se acompañará de las correspondientes normas escritas de higiene y uso.
6.º Cuando las actividades para las que se solicite la autorización impliquen el almacenamiento de las células o tejidos, se deberán especificar y definir las condiciones de almacenamiento en cada caso, incluyendo los márgenes de aquellos parámetros que son relevantes para el mantenimiento de las propiedades de las células o tejidos, como la temperatura, la humedad, o la calidad del aire.
7.º Las medidas de los parámetros que son críticos deben monitorizarse y controlarse y deben registrarse para poder demostrar que se cumplen las especificaciones de las condiciones de almacenamiento.
8.º Deberá disponerse de una infraestructura de almacenamiento que permita claramente separar y distinguir aquellos tejidos y células que están en cuarentena de aquellos que han sido rechazados, o de los que han sido aceptados y están disponibles, con objeto de impedir contaminaciones cruzadas y mezclas simples. Según el tejido o célula de que se hable se requerirán áreas físicamente separadas o un sistema de segregación seguro, pero en todo caso deberá especificarse el sistema y que con él se cumple la norma básica de preservar las condiciones biológicas y las de seguridad y calidad.
9.º Deberán existir procedimientos y normas para el acceso a los locales del establecimiento, para la circulación interna, para la higiene y mantenimiento, para la eliminación de basuras y material sucio o de desecho y para la restauración de todos los servicios tras una situación de emergencia. Estos procedimientos deberán ser objeto de control.
e) En cuanto a la documentación, su recogida y custodia:
1.º Todos los procedimientos operativos estandarizados para las actividades para las que se solicita la autorización deben estar adecuadamente registrados, y la documentación debe estar adecuadamente guardada y custodiada. Debe haber un sistema que garantice este punto. Los documentos deben revisarse periódicamente y los procedimientos deben estar siempre de acuerdo con las especificaciones básicas de este real decreto. El sistema asegurará la estandarización del trabajo y que todos las fases puedan ser objeto de rastreo: codificación, evaluación de las células o tejidos y del ambiente en su caso, obtención, procesamiento, preservación, almacenamiento, disponibilidad, transporte y distribución, y que se tienen en mente los aspectos relacionados con el control y gestión de la calidad.
2.º Los equipos, materiales o personal que estén involucrados en actividades críticas deben estar adecuadamente identificados y dicha identificación debe documentarse.
3.º Cualquier cambio en los documentos de los procedimientos operativos o de las normas de actuación debe ser revisado, fechado, documentado y puesto en marcha sin dilación por el personal autorizado para ello.
4.º Deberá existir un sistema que asegure la revisión periódica de los documentos, el registro de los cambios introducidos y que sólo las versiones actualizadas estarán en uso.
5.º La documentación debe quedar registrada de forma que sea una correcta representación de los resultados y que resulte fiable.
6.º Los registros deben ser legibles e indelebles, pueden estar en soporte papel o transferidos a cualquier otro sistema validado como un soporte informático o un microfilm.
f) En cuanto a los sistemas de calidad:
1.º Se desarrollarán auditorías con periodicidad no inferior a la bienal para la revisión de todas las actividades para las que se ha autorizado el establecimiento de tejidos. El objetivo de las auditorías es verificar que se trabaja de acuerdo a los protocolos aprobados y los requerimientos de este real decreto. Tanto los hallazgos como las medidas correctoras deben quedar documentados.
2.º El hallazgo de desviaciones de los estándares de calidad que se hayan establecido obligará al desarrollo de una investigación que debe quedar documentada y que incluya las decisiones o sugerencias sobre posibles medidas preventivas o correctoras.
3.º El destino de las células y tejidos que entran en la definición de «no conformidad» se decidirá de acuerdo con procedimientos previamente establecidos y supervisados por el responsable técnico y el responsable de área de calidad. Todas las células y tejidos afectados por no conformidades deber ser identificados y contabilizados.
4.º Las acciones correctoras deben iniciarse y completarse en el momento adecuado y de manera eficaz. Todas las acciones preventivas y correctoras estarán documentadas y se evaluarán en cuanto a su eficacia.
5.º El sistema de gestión de la calidad debe ser revisado y existirá un procedimiento que lo permita hacer y que tenga el objetivo de asegurar una mejora continua y sistemática del funcionamiento del establecimiento de tejidos.
3. Los requisitos para la autorización de actividades, procesamiento, almacenamiento y distribución de células y tejidos son:
a) En cuanto a procesamiento:
1.º Los procesos críticos de procesamiento de células y tejidos deben estar validados. En ningún caso las células y tejidos deben resultar peligrosas para el receptor o potencialmente ineficaces. Esta validación se basará en estudios realizados por el propio establecimiento de tejidos o en datos publicados o en la evaluación de los resultados clínicos de las células y tejidos que han sido distribuidos por el establecimiento en el caso de procedimientos de transformación o preparación de tejidos que están consolidados.
2.º Se debe demostrar que los procesos de validación se pueden llevar a cabo en el establecimiento de tejidos de forma efectiva y sistemática.
3.º Los procedimientos deben estar documentados como procedimientos operativos estandarizados, en adelante POE, y de acuerdo a lo establecido en el apartado 2.e), y debe asegurarse que las actividades se llevan a cabo de acuerdo con estos POE.
4.º Cuando vaya a aplicarse un procedimiento de inactivación microbiológica, éste debe estar documentado, especificado y validado.
5.º Antes de introducir cualquier cambio en las actividades de procesamiento, el proceso modificado debe validarse y documentarse.
6.º Las actividades de procesamiento deben evaluarse periódicamente, para asegurar que se cumplan los resultados deseados.
7.º Las actividades para descartar aquellas células y tejidos que no cumplen con los estándares requeridos deben realizarse de modo que se evite la contaminación de otras células y tejidos, del personal, del aparataje o del loca, así como la contaminación medioambiental.
b) En cuanto al almacenamiento:
1.º Debe haber un sistema de inventario de células y tejidos diseñado de forma que se asegure que no se pueden distribuir hasta tanto no hayan satisfecho todos los requerimientos de este real decreto. Debe haber un POE que detalla las circunstancias, responsabilidades y procedimientos para la liberación y posterior distribución de tejidos y células.
2.º Todos los procedimientos de almacenamiento se desarrollarán en condiciones controladas de forma que se garanticen las condiciones de credibilidad, calidad y seguridad de las células y tejidos.
3.º Existirán medios de control ambiental de las áreas de acondicionamiento y almacenamiento con el fin de evitar cualquier situación que pueda afectar negativamente a la funcionalidad, a la integridad o a las condiciones biológicas de células y tejidos.
4.º El tiempo máximo de almacenamiento se debe especificar para cada tipo de tejido o célula y condición de almacenamiento. El periodo determinado debe tener en cuenta entre otras cuestiones el posible deterioro de las propiedades de células y tejidos así como el uso al que van destinados.
5.º Deben poder identificarse claramente las células y tejidos almacenados en todas las fases del procesamiento en el establecimiento de tejidos y debe ser posible distinguir claramente entre aquellas células y tejidos que están en cuarentena y los que están listos para ser distribuidos o los que deben ser descartados.
6.º Los registros de información deben recoger claramente todos los datos relativos a la evolución y el procesamiento de forma que pueda demostrarse que se cumplan las especificaciones de este real decreto antes de liberar y distribuir las células y tejidos. Deben estar disponibles todos los datos médicos, los resultados de los tests de evaluación, los datos de procesamiento de todos los casos en los que era necesario un POE, debe asegurarse que éste se ha llevado a cabo con rigor por las personas autorizadas para ello, y también que están disponibles todos los registros de los controles de las condiciones de almacenamiento.
Si en algún momento, un sistema informático está encargado de liberar o facilitar cualquier resultado de un test de laboratorio o una validación, debe haber un sistema de rastreo que puede dar información al auditor sobre el responsable de liberar dicha información.
7.º El responsable técnico definido en el artículo 17 deberá aprobar un documento de evaluación de riesgos que determine el destino de todos los tejidos y células almacenados. Este documento tendrá en cuenta la evaluación del donante y los criterios de selección y aceptación de los resultados de los tests realizados, así como cualquier modificación de las fases de procesamiento que pueda mejorar la seguridad y la calidad de las células y tejidos.
c) En cuanto a su distribución y retirada:
1.º Se deben definir las condiciones y el tiempo máximo de transporte, que permitan mantener las propiedades biológicas y funcionales de las células y tejidos.
2.º El contenedor debe ser seguro y garantizar que las células y tejidos se mantienen en las condiciones especificadas. Todos los contenedores y recipientes de envasado deben estar validados para el objetivo que se persigue.
3.º Cuando la distribución la lleva a cabo un tercero debe haber un documento acordado de contrato que asegure que las condiciones de transporte requeridas se mantienen.
4.º Debe designarse el personal que, dentro del establecimiento de tejidos, está a cargo de la retirada, de indicar los motivos y la necesidad de retirar los productos y de iniciar y coordinar las actividades necesarias para ello.
5.º Debe haber un procedimiento documentado que fije el sistema para efectuar las retiradas, y que debe incluir una clara adscripción de responsabilidades y acciones que se deben iniciar. Se debe incluir la modificación correspondiente según el anexo VII.
6.º Las acciones deber ser emprendidas en tiempos predefinidos y deben incluir el rastreo y seguimiento de todos los tejidos o células que pueden estar implicados, con el objetivo de localizar a cualquier donante que pudiera haber contribuido a provocar una reacción adversa o efecto indeseado y retirar todos aquellos tejidos y células que hayan sido obtenidos de ese donante, así como notificarlo a los consignatarios y receptores de las células y tejidos de ese donante que puedan estar en riesgo.
7.º Debe haber procedimientos documentados para el manejo de las solicitudes de células y tejidos. Las reglas de distribución a los pacientes o los organismos o centros de implante deben estar documentadas y estar accesibles para los implicados en caso de requerirlo.
8.º Debe haber un sistema documentado para el manejo de los tejidos o células devueltos al establecimiento de tejidos, incluyendo los criterios de aceptación del inventario si ello es aplicable.
d) En cuanto al etiquetado:
1.º El establecimiento de tejidos debe disponer de un sistema de etiquetado que garantice que las etiquetas o los documentos cumplen con los siguientes requisitos de información:
i) El etiquetado del contenedor primario de las células/tejidos debe mostrar:
1) El número de identificación o código del tejido/célula, el tipo de células o tejidos y el lote cuando esto último proceda.
2) La identificación del establecimiento de tejidos.
3) La fecha de caducidad.
4) En el caso de que sea para uso autólogo, esto debe ir especificado: «para uso autólogo». Además, se mostrará el código de identificación del donante/receptor.
5) En el caso de donaciones dirigidas, se identificará el receptor.
6) Cuando se conozca que las células/tejidos son positivos para algún marcador de enfermedad infecciosa, deberán ir identificados como muestras de riesgo: «riesgo biológico».
Si, por razones de espacio, no es posible incluir la información referida en los puntos 4) y 5), ésta deberá ser facilitada en un documento añadido al contenedor primario. Dicho documento deberá ir embalado junto al contenedor primario de forma que se asegure que permanecen juntos.
ii) La información que se detalla a continuación puede figurar en la etiqueta o bien en un documento adjunto:
1) Descripción, definición y, si fuera relevante, las dimensiones del tejido o producto celular.
2) Morfología y datos funcionales cuando sea relevante.
3) Fecha de distribución de las células /tejidos.
3) Determinaciones biológicas que se han llevado a cabo en el donante y sus resultados.
4) Recomendaciones de almacenamiento.
5) Instrucciones para la apertura del contenedor, para el embalaje y para cualquier manipulación o reconstitución.
6) Fechas de caducidad después de la apertura o manipulación del contenedor.
7) Instrucciones para la comunicación de efectos o reacciones adversas.
8) Presencia de residuos potencialmente peligrosos (antibióticos, óxido de etileno, etc).
iii) Etiquetado externo para el contenedor de envío o transporte.
Para el envío, el contenedor primario debe ir incluido en un contenedor de transporte adecuadamente etiquetado. Esta etiqueta contendrá, al menos, la siguiente información:
1) Identificación del establecimiento de tejidos de origen, incluyendo la dirección, el teléfono y la persona de contacto.
2) Identificación del centro de implante de tejidos o establecimiento de tejidos destino, incluyendo la dirección, el teléfono y la persona de contacto.
3) La constatación de que el paquete contiene tejidos o células humanas y que debe ser manejado con cuidado.
4) Si se envían células vivas y el mantenimiento de la viabilidad es básico para el éxito del injerto, como es el caso de los progenitores hematopoyéticos, células precursoras, gametos o células embrionarias, debe añadirse en un lugar bien visible el anuncio de: «NO IRRADIAR».
5) Recomendaciones para las condiciones de transporte (posición, temperatura etc).
6) Instrucciones de seguridad.
7) Métodos de congelación o descongelación o cualquier otra manipulación necesaria cuando ello sea de aplicación.
4. Los requisitos específicos para optar a la autorización como centros, o unidades de implantación de tejidos humanos, según la actividad a desarrollar, son:
a) Actividades de implantación de progenitores hematopoyéticos, incluyéndose en ellos el implante de precursores hematopoyéticos procedentes de médula ósea, sangre periférica, cordón umbilical u otros.
1.º Se establecen como requisitos mínimos específicos comunes de los centros para obtener la autorización para los tres tipos de trasplante mencionados, los siguientes:
1) Disponer de personal facultativo especializado con experiencia acreditada en el trasplante de médula ósea.
2) Garantizar la disponibilidad de un médico con experiencia probada en el diagnóstico y tratamiento de las complicaciones del trasplante de médula ósea.
3) Disponer de personal de enfermería con formación en este tipo de cuidados.
4) Estar dotado de una Unidad de Cuidados Intensivos, de un Servicio de Diagnóstico por Imagen con disponibilidad de técnicas adecuadas y de laboratorios generales adecuados.
5) Disponer de un área de aislamiento antiinfeccioso adecuado.
6) Contar con un Servicio o Unidad de Hematología-Hemoterapia o Banco de Sangre, que será responsable del soporte hemoterápico adecuado, de la citoaféresis mecanizada y de la obtención, criopreservación y almacenamiento de los progenitores hematopoyéticos.
7) Disponer de un Servicio o Unidad de Farmacia y/o Nutrición capacitado para la elaboración de soluciones para nutrición entérica o parenteral ajustada a la situación de los pacientes.
8) Estar dotado de un Laboratorio de Anatomía Patológica que disponga de los medios técnicos y humanos necesarios para el diagnóstico de las complicaciones asociadas al trasplante y poder realizar los posibles estudios post-mortem.
9) Disponer de un Laboratorio de Microbiología donde se puedan efectuar los controles de las complicaciones infecciosas que presenten los pacientes.
2.º Dentro de este grupo de actividades y en función de los distintos tipos de trasplante para los que se solicite autorización, los centros tendrán que cumplir, además de todos los requisitos precedentes, los siguientes:
1) La autorización de los centros para realizar trasplantes alogénicos quedará condicionada a un número mínimo de procedimientos anuales que será determinado por la Comisión de Trasplantes y Medicina Regenerativa del Consejo Interterritorial del Sistema Nacional de Salud.
2) Para la realización de implantes alogénicos a partir de donantes familiares, el centro debe cumplir, además de los requisitos específicos comunes y los previstos en el apartado anterior, los siguientes:
i) Disponer de un laboratorio de histocompatibilidad, propio o concertado, con capacidad de determinar el polimorfismo del complejo principal de histocompatibilidad (MHC, HLA) para los loci A, B, C, DR y DQ en baja o alta resolución.
ii) Disponer de un área de aislamiento que como mínimo aplique un sistema de aislamiento invertido.
3) Para la realización de implantes alogénicos a partir de donantes no emparentados el centro, además de los anteriores requisitos (autoimplantes e implante de médula ósea a partir de donantes familiares) deberá garantizar la disponibilidad de un Laboratorio de Histocompatibilidad con capacidad de determinar los loci A, B, C, DRB1 y DQ por DNA de alta resolución.
b) Actividades de implante de tejidos osteo-tendinosos: disponer de una Unidad Quirúrgica especializada con al menos un especialista con experiencia demostrada en dichos trasplantes.
c) Actividades de implante de piel: disponer de una Unidad Quirúrgica especializada con al menos un especialista con experiencia demostrada en trasplante de piel.
d) Actividades de implante de válvulas cardíacas: disponer de una Unidad de Cirugía especializada, con amplia y reconocida experiencia en intervenciones con circulación extracorpórea, así como de, al menos, un profesional con experiencia demostrada en la implantación de válvulas.
e) Actividades de implantes de segmentos vasculares: disponer de una Unidad de Cirugía con al menos un especialista con experiencia en dichos trasplantes.
f) Actividades de implante de tejido ocular, incluyendo córneas, limbocorneal, esclera y otros tejidos oculares: disponer de una Unidad de Cirugía especializada con al menos un especialista con experiencia en dichos trasplantes.
g) Implantes de grupos celulares: disponer de la unidad y equipamientos necesarios para llevar a cabo los implantes específicos de que se trate.
1. Donantes fallecidos.
1.1 Criterios generales de exclusión.–Con carácter general, los posibles donantes que cumplan alguno de los criterios que se mencionen a continuación no se considerarán donantes válidos:
a) Causa de muerte desconocida, excepto en los casos en que se pueda realizar una autopsia y que ésta demuestre que no se encuentra en el cadáver ningún motivo de exclusión.
b) Historia de enfermedad no filiada.
c) Ingesta o exposición a algún tóxico que pueda ser transmitido, a dosis tóxicas, al receptor a través de los tejidos o células (cianuro, plomo, mercurio, oro, etc.).
d) Presencia o historia de enfermedad maligna, excepto el carcinoma primario basocelular, el carcinoma in situ de cervix uterino y algunos de los tumores primarios del sistema nervioso central en los que la evidencia científica nos dice que el riesgo de transmisión es aceptable desde el punto de vista de la seguridad y calidad. Los donantes con enfermedades malignas pueden ser aceptados como donantes de córnea, excepto en los casos de retinoblastoma, neoplasias hematológicas y otros tumores malignos que puedan afectar al polo anterior del ojo.
e) Riesgo de presentar enfermedades causadas por priones. Este riesgo incluye los siguientes ejemplos:
1.º Diagnóstico de enfermedad de Creutzfeld-Jakob no iatrogénica o variante de enfermedad de Creutzfeld‑Jakob o historia familiar de enfermedad de Creutzfeld-Jakob no iatrogénica.
2.º Historia de demencia rápidamente progresiva o enfermedad neurológica degenerativa de origen desconocido.
3.º Tratamiento previo con hormonas derivadas de la hipófisis (i.e. hormona del crecimiento). Receptores de duramadre, córnea o esclera. Personas sometidas a intervención quirúrgica no documentada donde pueda haberse utilizado duramadre.
f) Infección activa y no controlada en el momento de la donación, incluyendo infección bacteriana e infección sistémica viral parasitaria o fúngica, o infección localizada en los tejidos a utilizar. Los potenciales donantes con sepsis bacteriana pueden ser evaluados y considerados para la extracción de córneas si éstas se van a almacenar en cultivos que permitan la detección de contaminación bacteriana.
g) Historia, existencia de factores de riesgo de transmisión, evidencia clínica o tests de laboratorio positivos para HIV, hepatitis B, hepatitis C y HTLV I y II.
h) Historia de enfermedad autoinmune crónica que pueda haber dañado los tejidos a utilizar.
i) Presencia de otros factores de riesgo para trasmitir enfermedades, teniendo en cuenta la historia de viajes y la prevalencia local de enfermedades infecciosas.
j) Riesgo de que los tests biológicos puedan quedar invalidados:
1.º Por existencia de hemodilución (ver anexo III).
2.º Por tratamiento con inmunosupresores.
k) Presencia de signos físicos que puedan suponer un riesgo de transmisión de enfermedad.
l) Historia reciente de vacunación con virus atenuados, que puede constituir una fuente de contagio.
m) Receptores de xenotrasplante.
1.2 Criterios de exclusión específicos para la edad pediátrica.–Además de lo especificado en el punto anterior, que es igualmente aplicable a los donantes de edad pediátrica, cualquier niño nacido de madre portadora o enferma de VIH o que pueda incluirse dentro de los apartados del punto anterior debe ser excluido, salvo que se pueda demostrar que no existe riesgo de transmisión:
a) Los donantes menores de 18 meses nacidos de madres con marcadores positivos de VIH, hepatitis B o C o que tengan factores de riesgo para estas enfermedades, que hayan recibido lactancia materna en los 12 meses previos deben descartarse independientemente de los tests serológicos.
b) Los donantes menores de 18 meses nacidos de madres con marcadores positivos de VIH, hepatitis B o C que no han recibido lactancia materna en los 12 meses previos y que no presenten evidencia clínica ni historia compatible con haber estado infectados, cuyos tests serológicos sean negativos para VIH, hepatitis B o C pueden ser aceptados como donantes.
1.3 Examen físico externo.–Se debe realizar una exploración física detallada del cadáver para detectar si hay signos que puedan indicar que existe un riesgo de transmisión de enfermedad: tumores (i.e. melanoma), infecciones (i.e. úlceras genitales o condilomas anales), factores de riesgo de transmisión de enfermedad infecciosa (signos de venopunción, tatuajes o piercings no filiados).
2. Donante vivo.
2.1 Donante vivo autólogo.–El médico responsable del procedimiento terapéutico debe determinar, sobre la base de la historia clínica, la indicación terapéutica y la documentación disponible, la justificación para la donación y los criterios de seguridad.
Si las células o tejidos obtenidos van a ser almacenados, cultivados o sometidos a algún proceso de transformación «ex vivo» se realizarán los mismos tests biológicos que los requeridos para los donantes alogénicos. Los resultados positivos de cualquiera de los tests no impedirán el reimplante de las células, los tejidos o los productos derivados.
Ambos, paciente o su representante legal y el médico responsable, deben firmar el documento de donación con arreglo a las disposiciones legales vigentes y a lo establecido en el artículo 7. En dicho documento, el paciente deberá reconocer que la información que ha facilitado se ajusta a lo cierto dentro de su margen de conocimiento.
2.2 Donante vivo alogénico.–El donante se seleccionará sobre la base del conocimiento de su historia clínica y la entrevista personal realizada por el profesional médico responsable. Esta evaluación incluirá aquellos puntos que resulten relevantes en la identificación y selección de posibles donantes cuya donación pudiera representar un riesgo para la salud de terceros, como la posibilidad de la transmisión de enfermedades, o para su propia salud. En el caso de la donación de sangre de cordón umbilical o membrana amniótica, no deberá haber interferencia ni compromiso con el cuidado y la seguridad de la madre o el recién nacido.
Los criterios de selección de donantes vivos de tejidos o células para uso alógenico se establecerán y documentarán en el establecimiento de tejidos que los vaya a recibir, o en la unidad de trasplante, cuando se trate de una referencia directa de las células o tejidos del centro de obtención al de implante. Estos criterios incluirán los específicos de cada tejido o grupo celular más los que hagan referencia al estado general del donante, su historia clínica y de hábitos sociales, y los resultados de los tests de exploración clínica y de laboratorio designados para verificar el estado de salud del donante.
Se seguirán los mismos criterios generales de exclusión que se han especificado para los donantes fallecidos. En casos seleccionados de trasplantes de progenitores hematopoyéticos se podrán admitir donantes con marcadores virales B y C positivos. En los casos de donación de gametos dentro de la pareja se seguirán los criterios especificados al efecto (según el anexo IV).
Dependiendo del tejido o grupo celular se añadirán otros criterios de exclusión:
a) Embarazo: Excepto para la donación de progenitores hematopoyéticos y membrana amniótica.
b) Lactancia materna.
c) La posibilidad de transmitir enfermedades hereditarias en el caso de progenitores hematopoyéticos y gametos.
Ambos, el donante o su representante legal y el médico responsable, deben firmar el documento de donación con arreglo a las disposiciones legales vigentes y a lo establecido en el artículo 7. En dicho documento, el donante deberá reconocer que la información que ha facilitado se ajusta a lo cierto dentro de su margen de conocimiento.
1. Tests biológicos requeridos para los donantes.
1.1 Los siguientes tests se requerirán, como mínimo, en todos los casos de donación de células y tejidos:
HIV 1 y 2: Anticuerpos Anti HIV-1, 2.
Hepatitis B: HBs Ag. Anti. Hbc.
Hepatitis C: Anticuerpos AntiHVC (en casos de progenitores hematopoyéticos se requerirá además PCR).
Sífilis: Ver 1.4.
1.2 Los tests de anticuerpos Anti HTLV I y II se deberán realizar en aquellos donantes que viven o que vienen de zonas con una elevada incidencia de la enfermedad. También se realizarán en los donantes que sean parejas sexuales o hijos de personas que viven o vienen de zonas con elevada incidencia de la enfermedad.
1.3 Cuando el test de anticuerpos Anti HB-C sea positivo y el HBsAg negativo, será necesario realizar pruebas adicionales para determinar si los tejidos y/o células pueden ser utilizados o deben ser descartados.
1.4 En algunas circunstancias se realizarán tests adicionales dependiendo de la historia del donante o las características de las células o tejido a utilizar (CMV, T. cruzi, toxoplasma, malaria, Dengue, VEB, HLA, RhD).
1.5 Se aplicará un algoritmo diagnóstico para excluir la presencia de infección activa por Treponema Pallidum:
a) Test no reactivo, específico o no: permite la utilización de tejidos o células.
b) Test no específico reactivo: se debe realizar un test específico que, caso de ser no reactivo, permitirá la utilización de tejidos o células.
c) Test específico reactivo: se requiere una evaluación específica del riesgo para determinar el uso o no de las células y/o tejidos.
1.6 Para los donantes autólogos se tendrá en cuenta lo especificado en el anexo II punto 2.2.1.
2. Requerimientos generales de los tests biológicos.
2.1 Los tests se realizarán en laboratorios cualificados y autorizados por las autoridades competentes de la correspondiente comunidad autónoma. Se utilizarán kits con marcado CE, cuando estén disponibles en el mercado. El tipo de test utilizado deberá ser validado para el objetivo que persigue, de acuerdo al conocimiento científico y siguiendo las instrucciones del fabricante.
2.2 Los tests biológicos se realizarán en suero o plasma del donante. No deben realizarse en otros fluidos, como el humor vítreo o acuoso, salvo que esté específicamente justificado.
2.3 Cuando los donantes fallecidos han recibido transfusiones de sangre o componentes sanguíneos y/o coloides en las 48 horas precedentes al fallecimiento o cristaloides en la hora precedente al fallecimiento debe aplicarse el algoritmo del cálculo de la hemodilucción. Los establecimientos de tejidos podrán aceptar tejidos o células de donantes con tasas de hemodilución superiores al 50%, solo si los test de laboratorio están validados para muestras hemodiluídas o se encuentra alguna muestra de plasma o suero extraídas previamente a las transfusiones/infusiones.
a) Muestras de sangre «pre mortem»: si se han infundido componentes sanguíneos, sangre o coloides en las 48 horas precedentes a la toma de muestras o cristaloides en la hora precedente.
b) Muestras de sangre «post mortem»: si se han infundido componentes sanguíneos, sangre o coloides en las 48 horas precedentes al fallecimiento o cristaloides en la hora precedente al fallecimiento.
2.4 En el caso de donantes fallecidos, las muestras de sangre deben obtenerse antes del fallecimiento. De no ser así, las muestras se obtendrán lo antes posible, y, en todo caso, antes de transcurridas 24 horas desde el fallecimiento.
2.5 Otros supuestos:
a) En el caso de donantes vivos (excepto para células progenitoras de médula ósea y células progenitoras de sangre periférica, por razones de índole práctica), las muestras de sangre para serología se deben obtener en el momento de la donación o dentro del margen de los 7 días siguientes a la donación (del 0 al +7).
b) Cuando las células y/o tejidos se vayan a almacenar durante largos periodos, se requerirá una segunda determinación a los 180 días. En estos casos la «muestra de donación» se podrá tomar en el intervalo que transcurre entre los 7 días previos y los 30 días posteriores a la donación (del -7 al + 30).
c) Cuando las células y/o tejidos de un donante alogénico no se vayan a almacenar durante largos periodos y no se pueda proceder a la segunda determinación, se aplicará el punto a).
2.6 Si se aplican técnicas de amplificación de DNA para la determinación de la presencia de HIV, HBV y HLV, no se requerirá una segunda determinación. Tampoco será necesaria si durante la fase de procesamiento del tejido y/o grupo celular se incluye algún proceso validado de inactivación viral.
2.7 En el caso de donantes alogénicos de progenitores hematopoyéticos las muestras de sangre serán analizadas dentro del margen de 30 días pre extracción.
2.8 En el caso de progenitores hematopoyéticos de sangre de cordón umbilical se analizarán la sangre de la madre y la sangre del cordón.
2.9 En el caso de donantes neonatos, la muestra se obtendrá de la madre para evitar procedimientos médicos innecesarios para el niño.
1. Donación entre miembros de la pareja para su uso directo.
Los criterios de evaluación clínica o de laboratorio no se aplicarán a los casos de donación de células reproductoras entre miembros de una pareja para su uso directo.
2. Donación entre miembros de la pareja para su uso diferido.
Cuando las células reproductoras vayan a ser almacenadas o procesadas, se deberán cumplir los siguientes criterios:
2.1 El facultativo responsable del proceso de donación de gametos debe determinar y documentar, sobre la base de la historia clínica y la indicación terapéutica, la justificación para la obtención y los criterios de seguridad para la madre y los hijos que pudieran resultar del proceso.
2.2 Se realizarán los siguientes tests serológicos para evaluar el riesgo de contaminación cruzada:
HIV 1 y 2: Anticuerpos Anti HIV-1, 2.
Hepatitis B: Antígeno HBs / Anticuerpos anti HBC.
Hepatitis C: Anticuerpos Anti VHC.
Cuando los resultados de los tests para HIV 1 y 2 o de la Hepatitis B o C sean positivos o no estén disponibles, o cuando se sabe que el donante tiene algún factor de riesgo de transmisión de estas infecciones, se debe programar un sistema de almacenamiento aislado.
2.3 Los tests de determinación de anticuerpos anti HTLV I y II se realizarán en donantes que viven o vienen de áreas con una elevada incidencia de enfermedad o cuyas parejas sexuales o progenitores vengan o vivan en áreas de elevada incidencia de enfermedad.
2.4. En algunas circunstancias se requerirán tests adicionales (malaria, toxoplasma, Tripanosoma cruzi, dengue, CMV, VEB, RhD) dependiendo de la existencia de viajes, o exposición a riesgo de contagio, o de las características de las células obtenidas.
2.5 El hecho de que los tests sean positivos no impide necesariamente que se puedan utilizar las células obtenidas, o los productos derivados, en casos de donación entre personas de la misma pareja, siempre de acuerdo a la normativa vigente.
2.6 Cuando el test de HIV 1 y 2 o de la hepatitis B o C sean positivos o no se disponga de los resultados, o cuando el donante presente algún criterio de riesgo de infección, se utilizará un sistema de almacenamiento aislado.
3. Donaciones fuera de la pareja.
El uso de células reproductoras de donantes diferentes a la pareja habitual deberá cumplir los siguientes criterios:
a) Los donantes se seleccionarán sobre la base de su historia clínica, que debe hacer el facultativo responsable. Esta evaluación incluirá cualquier factor que pueda resultar relevante en la identificación y selección de aquellas personas cuya donación pueda representar un riesgo para la salud de terceros, como la posibilidad de transmitir una enfermedad, o para sí mismos (i.e. inducción y/o estimulación de la ovulación, sedación, riesgos asociados a la extracción de óvulos o consecuencias de índole psicológica).
b) Los donantes deben tener marcadores serológicos negativos para HIV 1 y 2, HVC y HVB y sífilis. Los donantes de esperma deben tener, además, marcadores negativos para chlamidia en una muestra de orina y por determinación mediante PCR.
c) Se realizarán tests de determinación de anticuerpos anti HTLV I y II en aquellos donantes que viven o provienen de zonas con elevada incidencia de enfermedad o cuyas parejas sexuales o progenitores viven o provienen de áreas con elevada incidencia de la enfermedad.
d) En algunas circunstancias se requerirán tests adicionales dependiendo de la historia clínica del donante o de las características de las células o tejidos (i.e. malaria -CMV-Tripanosoma cruzi, RhD).
e) En el caso de donaciones autólogas se aplicará lo establecido en el anexo II punto 2.1.
f) Se llevará a cabo una evaluación de la carga genética en relación a la existencia de genes autonómicos recesivos de acuerdo al conocimiento científico y a la prevalencia conocida en la etnia del donante.
g) Se llevará a cabo una evaluación del riesgo de transmisión de enfermedades hereditarias conocidas y presentes en la familia. Se informará a los implicados de los resultados obtenidos de acuerdo con lo dispuesto en la Ley 41/2002, de 14 de noviembre, básica reguladora de la autonomía del paciente y de derechos y obligaciones en materia del información y documentación clínica y la Ley 14/2006, de 26 de mayo, sobre técnicas de reproducción humana asistida. Esta información deberá ser lo más completa posible en relación a los riesgos asociados y a las medidas adoptadas o que se puedan adoptar, y debe ser transmitida y explicada claramente al receptor.
h) Requerimientos básicos para la realización de tests biológicos.
Los test biológicos se realizarán de acuerdo con lo especificado en el punto 3.2 del anexo III:
1.º Las muestras de sangre se obtendrán en el momento de la donación.
2.º Las muestras de esperma se mantendrán en cuarentena, al menos, 180 días, tras lo cual se repetirán los tests biológicos. Esta segunda evaluación se podrá evitar si la primera determinación se hizo mediante test de amplificación de ácidos nucleicos. Igualmente, se podrá evitar la segunda determinación de tests biológicos si en el proceso de transformación o manejo posterior las células van a sufrir un proceso validado de inactivación viral.
1. Donación y extracción.
1.1 Consentimiento e identificación del donante.-Antes de proceder a la extracción de las células y tejidos, el responsable del procedimiento o persona autorizada para ello, debe confirmar y registrar:
a) Que el consentimiento para la extracción se ha obtenido conforme a lo establecido en la legislación vigente.
b) Cómo se ha realizado la identificación del donante.
c) Que, en el caso de donaciones de vivo, el donante ha entendido la información facilitada, ha tenido la oportunidad de preguntar sus dudas y ha obtenido respuestas satisfactorias y que ha confirmado que la información que ha facilitado, con respecto a su historial clínico, es cierta hasta donde llega su conocimiento.
1.2 Evaluación del donante.
1.2.1 El responsable del procedimiento de extracción o persona autorizada para ello debe recoger y registrar toda la información clínica y social del donante que resulte relevante para la evaluación tal y como se describe en el apartado 1.4 de este anexo (documentación del donante).
1.2.2 En el caso de los donantes vivos se llevará a cabo una entrevista personal durante la cual se completará un cuestionario estructurado. En el caso de los donantes fallecidos el cuestionario se rellenará con la ayuda de:
a) La familia o los allegados (anamnesis social y de hábitos).
b) El médico que le ha tratado.
c) Su médico de cabecera (si procede).
d) El historial médico / los resultados de la autopsia.
1.2.3 Se llevará a cabo una exploración física del donante para detectar aquellos signos o marcas que puedan ser sospechosos de transmisión de enfermedad o resulten complementarios a la información de la historia clínica y que puedan obligar a evaluaciones adicionales antes de aceptar el donante: tumores (melanomas), infecciones (úlceras genitales o condilomas anales), factores de riesgo para padecer enfermedades transmisibles (venopunciones), traumatismos o cicatrices de operaciones recientes o antiguas.
1.3 Extracción de células y tejidos.
1.3.1 Los procedimientos de extracción serán los adecuados para el tipo de donante y el tipo de células o tejidos que se van a obtener, así como para garantizar la protección del donante.
1.3.2 Los procedimientos utilizados deberán garantizar que se protegen las propiedades de estas células o tejidos que se requieren para su uso clínico, y que minimizan los riesgos de contaminación microbiológica, especialmente si las células o tejidos no van a ser objeto de tratamiento de esterilización subsiguiente.
1.3.3 En el caso de donantes fallecidos, se debe registrar el lugar de la extracción (o se debe describir en el informe de extracción) que deberá ser un área restringida. El equipo médico que vaya a realizar la extracción deberá adoptar las medidas preventivas de contaminación más adecuadas en cada caso. En general, ello debe incluir el lavado de las superficies de trabajo con soluciones antisépticas, la preparación de un campo estéril, realizar un lavado quirúrgico de manos y utilizar guantes y batas estériles, así como mascarilla y gorro.
1.3.4 En el caso de las extracciones de donantes fallecidos se debe registrar la hora del fallecimiento y la de la extracción, registrándose el intervalo y asegurándose que no se exceden los límites que garantizan que se preservarán las características y propiedades biológicas de las células y tejidos.
1.3.5 Una vez se hayan extraído los tejidos o grupos celulares de un donante fallecido, se llevará a cabo una reconstrucción de las zonas afectadas, de manera que se acerque lo más posible a su apariencia anatómica previa.
1.3.6 Cualquier suceso que ocurra durante el procedimiento de extracción y que pueda resultar o haya resultado perjudicial para el donante, así como cualquier investigación adicional derivada de estos hechos para determinar su causa, debe ser adecuadamente recogida y evaluada.
1.3.7 Deberá haber guías de procedimiento estandarizado disponibles para minimizar el riesgo de contaminación por parte de miembros del staff que pudieran estar infectados con enfermedades transmisibles.
1.3.8 Para la extracción de células y tejidos se utilizaran instrumentos y sistemas estériles de alta calidad, validados y/o específicamente certificados y mantenidos regularmente para el uso al que están destinados.
1.3.9 Cuando se utilice instrumental de múltiple uso, deberá haber procedimientos estandarizados validados disponibles para la limpieza y esterilización de dicho material.
1.3.10 Siempre que sea posible se utilizarán materiales con certificación UE y se entrenará adecuadamente al staff implicado para el manejo de dicho instrumental.
1.4 Documentación del donante.
1.4.1 Para cada donante deberá prepararse un fichero que contenga:
a) Identificación del donante (nombre, apellidos y fecha de nacimiento con su equivalente identificativo).
b) En el caso de donaciones de neonatos o sangre de cordón o cualquier otro tejido o grupo celular obtenido en el momento del parto, se registrarán el nombre y fecha de nacimiento de la madre, la fecha de nacimiento del donante y su nombre si se conoce.
c) Sexo, edad, historial médico y social.
d) Resumen de la exploración física.
e) Fórmula del cálculo de hemodilución (si procede).
f) Documento de consentimiento para la obtención.
g) Datos clínicos, resultados de los tests de laboratorio y cualquier otra determinación o pruebas realizadas.
h) Resultado del informe si se ha procedido a un examen necrópsico.
i) En el caso de los progenitores hematopoyéticos se registrará la documentación relativa a la idoneidad del donante para un determinado receptor.
1.4.2 El equipo de extracción elaborará un informe del procedimiento de extracción del cual se enviará una copia al establecimiento de procesamiento de tejidos. En este informe se recogerá, al menos, la siguiente información:
a) Identificación, nombre y dirección del establecimiento de destino que va a recibir el grupo celular y/o tejidos extraídos.
b) Identificación del donante, incluyendo cómo se llevado a cabo la identificación y quién lo hizo.
c) Causa, fecha y hora de la muerte (en donante fallecido).
d) Descripción e identificación de los tejidos y células extraídos y de las muestras obtenidas para la evaluación.
e) Identificación del responsable del grupo de extracción y firma del mismo.
f) Fecha y hora (de comienzo y finalización), lugar de la extracción y procedimiento utilizado (POE, si procede). Descripción del área y las condiciones en que se realizó la extracción (si procede).
g) Incidentes ocurridos durante la extracción.
h) En el caso de donantes fallecidos, información sobre los métodos y condiciones de la conservación del cadáver: si ha estado refrigerado o no, temperatura, tiempo, comienzo y fin de la refrigeración.
i) Reactivos y soluciones de conservación utilizadas (Identificación de lotes).
j) En el caso de donantes de esperma la información mínima a consignar será:
1.º Nombre del establecimiento de tejidos de destino.
2.º Datos de identificación del donante.
3.º Fecha y hora de la obtención.
k) La información relativa al donante deberá ser archivada y protegida contra modificaciones no autorizadas, custodiada de forma apropiada y accesible para la autoridad competente, al menos hasta 30 años después del uso clínico o caducidad de las células o tejidos obtenidos.
1.5 Empaquetado.
1.5.1 Tras la extracción todas las células y tejidos serán empaquetados de forma que se minimicen los riesgos de contaminación y se asegure la temperatura requerida para preservar las características y propiedades biológicas y funcionales de las células y tejidos.
1.5.2 Las células y tejidos empaquetados deberán ser transportados en contenedores adecuados para el transporte de material biológico y que mantenga su calidad y seguridad.
1.5.3 Las muestras de tejido o sangre que acompañen a las obtenidas para uso último con el fin de servir para ulteriores tests o determinaciones analíticas, deberán ir adecuadamente etiquetadas. En estas etiquetas debe figurar la identificación del donante y la información relativa al lugar y el momento en que se recogió el espécimen.
1.6 Etiquetado de los tejidos o células extraídos.
1.6.1 En los contenedores internos de células y/o tejidos para uso humano debe figurar una etiqueta que contenga, al menos, la siguiente información:
a) Código de identificación del donante.
b) Tipo de célula y/o tejido.
1.6.2 En el caso de que el contenedor lo permita, en virtud de sus dimensiones, deberá figurar además:
a) Fecha y hora de la obtención.
b) Precauciones (si procede).
c) Aditivos utilizados (si procede).
d) En caso de donaciones directas debe identificarse el receptor.
e) En caso de donaciones autólogas deberá figurar: «Sólo para uso autólogo».
1.7 Etiquetado del contenedor externo de transporte.–En el contenedor de transporte de las células y/o tejidos debe figurar una etiqueta donde se especifique la siguiente información:
a) «Muestra biológica de células/tejidos-Manejar con cuidado».
b) Identificación del establecimiento de tejidos de origen del tejido y/o grupo celular, incluyendo la dirección y el teléfono y la persona de contacto para cualquier contingencia.
c) Identificación del establecimiento de tejidos de destino, incluyendo la dirección y el teléfono, así como la persona de contacto a quien hay que entregar el contenedor.
d) Fecha y hora de inicio del transporte.
e) Especificaciones para mantener las características biológicas de las células o tejidos durante el transporte (si procede).
f) Especificaciones de almacenamiento si procede (i.e. NO CONGELAR).
g) En caso de que los tejidos o células puedan verse afectados por los rayos X debe figurar claramente «NO IRRADIAR».
h) En casos de productos que se conoce que son potencialmente contaminantes o de los que se desconocen los resultados de los tests serológicos debe especificarse: «RIESGO DE CONTAMINACIÓN BIOLÓGICA».
i) En el caso de donaciones autólogas debe figurar claramente «Para uso autólogo exclusivamente».
2. Recepción del tejido y/o grupo celular en el establecimiento de tejidos.
2.1 Condiciones generales.–Cuando el tejido y/o grupo celular extraído llegue al establecimiento de tejidos, se llevará a cabo un procedimiento documentado de verificación de que el envío recibido cumple con todos los requisitos exigidos, tanto en este real decreto como en las especificaciones del propio establecimiento de tejidos, en relación a las condiciones de transporte, de empaquetado y de etiquetado, y en relación a las muestras para ulteriores controles e información y documentación que deben acompañar a los tejidos y/o células.
El establecimiento de tejidos debe asegurar que los tejidos y/o células recibidos permanecen en cuarentena hasta que ellos mismos y toda la documentación acompañante haya sido objeto de los análisis, controles inspecciones o verificaciones requeridos en este real decreto y en las especificaciones del propio establecimiento. La revisión de la documentación, así como la consiguiente decisión sobre su aceptación, debe ser hecha por la persona autorizada o designada en el establecimiento de tejidos.
Cada establecimiento de tejidos debe tener un procedimiento documentado para asegurar que los envíos de tejidos y/o células recibidos que no cumplen con los requisitos establecidos, o cuya documentación está incompleta o que están a la espera de completar los resultados de la evaluación del donante, se almacenan de forma que no haya riesgo de contaminación para otros tejidos y/o células preservados, almacenados o procesados en el mismo establecimiento.
2.2 Registro de datos.–Los datos que se deben registrar en el establecimiento de tejidos (excepto en el caso de la donación de células reproductoras entre miembros de la pareja) serán, al menos, los siguientes:
a) Consentimiento o autorización para la extracción, donde se consigne el propósito de utilización (uso terapéutico o investigación o ambos) y cualquier instrucción específica para su destrucción cuando no se utilicen para el propósito con el que se obtuvieron.
b) Los relativos a la identificación del donante y sus características, incluyendo el tipo de donante y la causa de muerte, si procede, tal y como se ha descrito en la sección: «Documentación».
c) Los relativos a la historia clínica del donante y al procedimiento de extracción, tal y como se ha reseñado en el anexo correspondiente.
d) Los relativos a la exploración física del donante, los resultados de los tests de laboratorio o de cualquier otra prueba practicada al donante, incluyendo los de la necropsia en caso de haberse realizado.
e) El informe completo de evaluación del donante firmado por el responsable del proceso de evaluación y selección o persona autorizada.
f) Los relativos al procedimiento de extracción, tal y como se recoge en el anexo correspondiente, incluyendo el lugar de la extracción y la persona responsable.
g) Los tejidos y/o células que se reciben y sus características.
h) En caso de tejido autólogo, es necesario especificar además:
1.º Las características de la lesión o proceso patológico que se va a tratar.
2.º Alergias medicamentosas o a productos que puedan ser utilizados en la conservación y procesamiento.
2.2.1 En el caso de cultivos celulares para uso autólogo se consignará además la información sobre posibles alergias del receptor (i.e. antibióticos).
2.2.2 En el caso de las donaciones de células reproductoras fuera de la pareja habitual, se consignarán además los siguientes datos relativos a los donantes:
talla, peso, raza, color de piel (pálido, moreno), color de ojos (marrón, verde, ámbar, azul, negro), color de pelo (rubio, castaño claro, castaño oscuro, pelirrojo, negro), textura de pelo (liso, ondulado, rizado), grupo sanguíneo y Rh.
2.2.3 En el caso de células reproductoras que van a ser utilizadas en el seno de la pareja habitual, los datos que se consignarán son:
a) Consentimiento/autorización para la extracción, donde se consigne el propósito de utilización (uso terapéutico/investigación o ambos) y cualquier instrucción específica para su destrucción cuando no se utilicen para el propósito con el que se obtuvieron.
b) Datos de identificación del donante: tipo de donante, edad, sexo, presencia de factores de riesgo y causa de la muerte en caso de donantes fallecidos.
c) Datos de identificación de la pareja:, edad, sexo y presencia de factores de riesgo.
d) Lugar de la obtención del grupo celular.
e) Células o tejidos obtenidos y sus características más relevantes.
3. Requerimientos para la distribución directa al centro de implante de tejidos y/o células específicos.
Excepcionalmente la unidad de Coordinación Autonómica de Trasplantes y/o la ONT podrá autorizar el envío directo de algunas células o tejidos específicos desde el centro donde se realiza la extracción al centro de implante para su uso inmediato. (i.e. células progenitoras hematopoyéticas, córneas, etc.). En todo caso se exigirán los requisitos especificados en estos anexos en cuanto a la identificación, extracción, empaquetado, envío, preservación y etiquetado.
1. Información que debe guardar y custodiar el establecimiento de tejidos:
a) Identificación del centro, unidad u organismo de obtención autorizado.
b) Número identificativo único de donación.
c) Fecha de obtención.
d) Lugar de obtención.
e) Tipo de donación/obtención:
1.º Fallecido –Vivo.
2.º Multitejido –Tejido/grupo celular único.
3.º Alogénico –Autólogo.
f) Identificación del establecimiento de tejidos.
g) Tipo de tejido o grupo celular.
h) Número de lote, si procede.
i) Número de subpartición, si procede.
j) Fecha de caducidad.
k) Estatus del tejido/grupo celular:
1.º Disponible.
2.º Descartado.
3.º Cuarentena.
l) Descripción del producto células o tejido: origen, fases de procesamiento o transformación aplicadas, materiales y aditivos que han estado o están en contacto con las células o tejidos y que pueden afectar a su calidad y/o seguridad o cuya presencia debe tenerse en cuenta por razones de seguridad para las personas en las que se apliquen (ie. Presencia de antibióticos y posibles reacciones alérgicas).
m) Etiquetado interno y externo del tejido o grupo celular.
n) Fecha de disponibilidad.
ñ) Identificación del centro o unidad de aplicación.
2. Información que debe guardar y custodiar el centro o unidad de aplicación:
a) Identificación del establecimiento de tejido proveedor.
b) Identificación del responsable de la unidad o centro de aplicación.
c) Tipo de tejido/grupo celular.
d) Identificación del producto.
e) Identificación del receptor o persona en la que se aplica el tejido o grupo celular.
f) Fecha de utilización, aplicación o en su caso descarte y causa de no utilización en este último supuesto.
Información contenida
1. Identificación de la donación:
a) Identificación de la donación.
b) Identificación del establecimiento de tejidos.
2. Identificación del producto:
a) Código de producto básico.
b) Número de partición.
c) Fecha de caducidad.




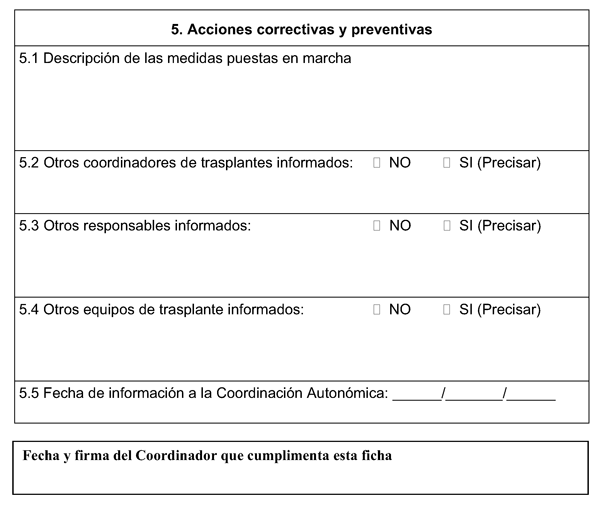
Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid




